【序】医薬の開発において、リード化合物の役割は極めて重要である。しかし、これまでリード創製は偶然やrandom screeningなどに頼ることが多く、医薬開発の障害となってきた。ますます高度化する社会のニーズに応え、優れた医薬を効率よく創り出していくためには、従来の方法に加えて新たな論理的方法の確立が不可欠である。そこで、本研究では偶然や試行錯誤に頼らないリード創製手法を可能にすることを目的として、活性に必要な三次元的特徴を備えた分子骨格をコンピュータに構築させる方法論の開発を行った。本方法は受容体の立体構造が既知・未知のいずれの場合にも適用できる方法であり、いくつかの系に適用してその有効性を確認した。 【方法】薬物分子と受容体の間の認識には、ファンデルワールス相互作用・静電相互作用・水素結合・疎水相互作用などが重要な役割を果たしており、一つの受容体に安定に結合できるリガンド構造としては多様な分子骨格が可能である。これらの構造には活性に必須と推定される複数の官能基が存在するが、活性発現にはこれらの官能基の有無だけではなく、それらの位置関係が適切に保たれていることが重要である。そこで、これらの官能基の位置と方向を保ち、一定の分子形状にそれらを含む多数の安定な構造をコンピュータに提示させることができれば、それらの構造を適切に選択・構造修正することにより、既知薬物とは異なった骨格のリードとなりうる分子が設計できると考えられる。このような基本的考えに基づき、リード化合物の候補となる分子構造を構築するためのアルゴリズムを開発し、ソフトウェア化した。方法の流れを図1に、分子構造構築の中心となる官能基間の結合経路探索法を模式的に図2に示す。 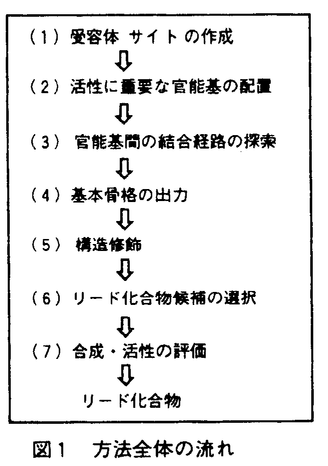 図1 方法全体の流れ 図1 方法全体の流れ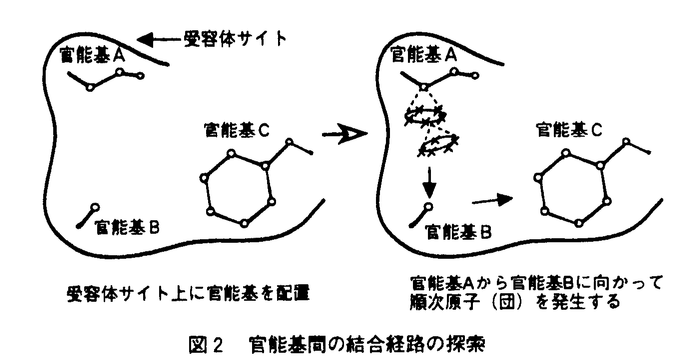 図2 官能基間の結合経路の探索 図2 官能基間の結合経路の探索 (1)新規分子構造構築の場として、受容体の形状・物理化学的性質を三次元格子点上に展開した受容体サイトを準備する。受容体サイトは、受容体の立体構造が既知の場合には、薬物結合部位に受容体全原子の及ぼす諸々のポテンシャルを記述することにより、また受容体の立体構造が未知の場合には、単独分子あるいは適切に分子重ね合わせを行った複数の既知薬物分子からレセプターマッピングを行うことにより導かれる。 (2)この受容体サイト上に活性に重要と推定される官能基を複数配置する。 (3)指定した官能基の位置と方向が保存されるように、受容体の形状・物理化学的性質等を考慮しながら官能基間を結び得る原子(団)の組み合わせを幅広ぐ探索する。この際、原子(団)の間はそれぞれの結合に標準的な結合長・結合角を保持し、エネルギー的に無理のないねじれ角を多数選ぶ。 (4)官能基間が種々の原子(団)で結ばれた、多様な基本骨格が出力される。 (5)必要に応じて環構造・水素結合性官能基・疎水性官能基の追加や構成原子の入れ替え等の基本骨格の修飾を行い、受容体とより良好な相互作用が行えるようにする。 (6)分子軌道計算・分子力学計算・分子動力学計算等を駆使して出力コンフォマーが十分安定で、受容体との良好な相互作用を維持できるかどうか検討し、少数にしぼりこむ。さらに合成化学的な見地からも構造修飾を加えて選択する。 (7)合成・活性の評価を行う。 《適用例1》受容体の立体構造が既知の系としてHIV-I protease(HIV-PR)に適用し、非ぺプチド性阻害剤を設計することを試みた。HIV-PRはaspartic acid proteaseの一種で、homo dimerとして働き、gag-pol前駆体からHIVが成熟したvirusとなるのに必要な蛋白質を切り出す機能を持っている。そこでHIV-PRを効率よく阻害することによりvirusの増殖を抑えることができる。X線結晶解析により明らかにされている酵素の活性部位と既知のぺプチド性阻害剤の構造を図3に示す。活性中心の水と阻害剤の水素結合、S2〜S2’の疎水性ポケットに収まる疎水性の官能基、Asp25との水素結合が活性に重要と考えられ、この酵素活性部位の構造をもとに受容体サイトを準備した。本方法で配置する官能基とその位置は必ずしも既知リガンドの官能基に固定されないが、本研究では活性中心の水と水素結合できる既知リガンドのカルボニルを受容体サイト上に配置して分子構築を行い、多数の構造を出力した。これらの構造にAsp25と水素結合しうる水酸基と上記の疎水性部位と相互作用しうる疎水性の基を付け加え分子を完成させた。さらに、これらの出力構造が安定コンフォマーとして存在しうるかどうかを検討するため高温分子動力学計算により可能なコンフォマーを幅広く探索し、受容体中でのエネルギー極小化計算を行った。その結果、次の条件を満たすものをリード化合物の候補構造とすることにした。(1)受容体中で比較的低エネルギーのコンフォマーで存在し得る。(2)活性中心の水およびASp25と良好な水素結合を保てる。(3)S2〜S2’の4つの疎水性部位のうちの少なくとも3つ以上を疎水性の基が占めている、等である。これらの候補構造についてさらに合成化学的見地からの検討も行い、合成を試みるべき化合物を決定した。その構造と酵素阻害アッセイの結果を併せて図4に示す。ぺプチド性の既知阻害剤と比しては非常に弱い活性しか認められなかったが、数少ない非ペプチド性阻害剤であるhaloperidolとは同等の阻害活性を持つことが示された。今後、適切な構造修飾により10〜1000倍程度の活性の増強は可能と思われる。 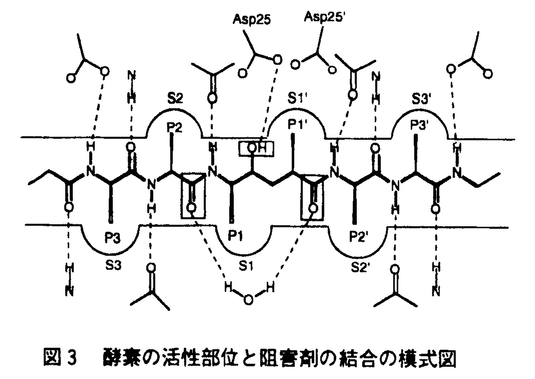 図3 酵素の活性部位と阻害剤の結合の模式図 図3 酵素の活性部位と阻害剤の結合の模式図 図4 合成された化合物と酵素阻害アッセイの結果 図4 合成された化合物と酵素阻害アッセイの結果 《適用例2》薬物受容体の立体構造が知られていない場合の例として古くからよく研究されているモルヒネ系鎮痛活性化合物の系を選んで方法論の有効性をテストした。モルヒネ(図5)の鎮痛活性にはベンゼン環とプロトネートした三級窒素の存在が必要と考えられている。そこでモルヒネの結晶構造をもとに受容体サイトを構築し、これらの官能基の位置と方向を保存しつつ、sp3炭素のみを用いて可能な構造を構築させた。プログラムには有機化合物に必要な原子種が幅広く用意されているが、本研究ではモルヒネ骨格の再現のテストを目的としてsp3炭素のみを用いた。これら多数の出力構造を最適化した後、コンフォメーション的安定性・官能基間の位置関係・受容体サイトへのフィットの良さ等の観点から有望な分子の選択を行った。この中には例えば1から6(図6)のようにモルヒネと同等の活性を持つ既知薬物分子の骨格が多数含まれていた。またモルヒネを含む複数のオビオイド活性分子の重ね合わせにより構築した受容体サイトからも構造構築を行い、上記モルヒネ単独分子からの出力構造と比較した。その結果、さらに多くの既知活性構造が出力構造中に含まれ(7〜9;図6)、必須官能基や活性コンフォメーションの推定だけでなく、新規活性構造構築にも分子重ね合わせが有用なことを示した。新規リードとして有望な構造としてはすべての条件をよく満たす10(図6)が選択できるが、合成するには至っていない。  図5 モルヒネ 図5 モルヒネ 図6 出力された構造の例 図6 出力された構造の例 【選択基準】客観的な数値的選択基準の設定が次なる重要課題であるが、それには薬物・受容体を含めた系全体の自由エネルギーから受容体への結合の強さを予測する必要がある。そうした目的には溶媒和や疎水相互作用についての理論的研究が不十分であり、現在の計算化学的手法で正しく見積もることはできない。またエンタルピーだけではなくエントロピーの寄与が重要であり、それらを考慮する方法論の開発が必須である。なお、本研究で考慮したのは受容体への結合であり、作用点への到達については別途考慮する必要がある。 【まとめ】論理的なリード創製手法の確立を目的として、活性に必須と思われる3次元的特徴を満たす多数の構造を自動構築する方法の開発に成功した。HIV-PR阻害剤の系においては、本方法により出力された構造をもとに選択・設計した化合物の活性を実験的に証明できた。モルヒネ系鎮痛化合物の設計については、本方法により出力された構造中にはモルヒネと同等の鎮痛活性がすでに確認されている種々の化合物の基本骨格が多数含まれていた。これらの結果から、本方法の基本概念の正しさとリード創製への有用性が示されたといえる。 |