| 内容要旨 | | 蛋白質の三次元構造がほぼ同じであると推定できるのは、通常、一次構造の相同性が25%以上ある場合とされている。一方、X線結晶解析などによって蓄積されてきた蛋白質の三次元構造をアミノ酸配列と比較すると、相同性が25%以下にもかかわらず、相互によく似た三次元構造をとるものが多く存在する。それらは、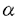 ヘリックス、 ヘリックス、 シート、ターン等の二次構造要素が相互に組み合わさったドメイン構造に多く見られる。 シート、ターン等の二次構造要素が相互に組み合わさったドメイン構造に多く見られる。 Triose phosphate isomerase(TIM)に代表される平行鎖とヘリックスが交互に8回並ぶ樽状のドメイン構造をもつ/バレル蛋白質の場合には、一次構造の相同性を示すファミリーに分類できるが、ファミリー間の相同性は25%以下と低い。本研究では、/バレル蛋白質においてどの領域がへリックスと鎖となるかを予測し、更にそれらの配置を立体的に配列させるドメイン構造予測法の開発を目的としている。この手法では、三次元構造既知のaldolaseやTIMなど16種類のファミリーに属する蛋白質とそれぞれ相同性の高い蛋白質の一次構造を統計的に解析する。ついで、20種類の構造既知の蛋白質について、それ自身のファミリーのデータを含まない統計データに基づいて二次構造要素の配置のシミュレーションを行う。開発したこの方法により、三次元構造が未知の蛋白質の一次構造データから、/バレル構造を正しく抽出することが可能となった。 方法 図1に示すような手順により/バレル構造を構成する二次構造要素の領域をまず選び出す。  図1./バレル構造予測の手順 図1./バレル構造予測の手順 1)/モチーフの候補選出シミュレーション /バレルの構造予測では、どの領域がヘリックス、ターン及び鎖から成る「/モチーフ」に成り得るかを正しく予測しなければならない。そのため、次の2段階の方法を採る。 a)/モチーフ関数M:/モチーフ中の鎖が連続する7残基のアミノ酸残基から構成されると仮定し、そのN端側5残基とさらにN端側にあるヘリックス部分に相当する8残基分の配列、合計20残基の配列中にどのようなアミノ酸残基(singlet)が出現しているかを検索する。これにより、疎水性アミノ酸残基、親水性アミノ酸残基などの位置によって異なる出現頻度が得られる。また、異なる位置のアミノ酸残基同士の組み合わせ(doublet)の出現頻度を求める。これらの統計データを基に、実際に/モチーフを形成している領域に対する値が、それ以外の領域に対する値よりもできるだけ高くなるような/モチーフ関数Mを定める。  上式において、s(i)はiの位置におけるアミノ酸残基の出現頻度値、d(i,i’)はiとi’の位置におけるアミノ酸残基の組み合わせの出現頻度値である。/モチーフ領域のM値ができるだけ大きくなるようにs(i)、d(i,i’)に対応した重みWs(i)、Wd(i,i’)を求める。 b)triplet消去法:/バレル蛋白質の/モチーフの場合、何残基も離れた別のモチーフと相互作用して構造を形成するため、M値の高いものと低いものが現われ、モチーフ候補の数が100本以上と多くなる。更に候補を絞るために、3残基のアミノ酸残基の組み合わせ(triplet)の寄与も加える。tripletの場合、一次構造のデータが不足しており、tripletの出現頻度を求めることが困難であるため、tripletの寄与を関数Mに加えることは出来ない。そこで、異なるファミリーに類似したtripletがある場合に/モチーフの候補として抽出する方法を試みる。同一のアミノ酸残基同士の類似度を1.00、イソロイシンとロイシンのように物理化学的性質の近い残基同士の類似度を0.96というように1.00に近い値とし、性質の似ていない残基同士の値を0.00に近い値に定める。3残基の類似度の平均値をtriplet間の類似度Rとする。/モチーフ毎に異なるファミリーの一次構造データベースから最高のRをとるtripletを検索する。全ての/モチーフを候補として抽出できるRの下限値Rminを求め、Rminよりも低いR値をとるtripletをもつ領域をモチーフの候補からはずすことにする。20残基から成るモチーフ内の可能なtriplet.の組み合わせの総数(20×19×18)/(3×2×1)=1140組のRminを解析すると、この値は0.646〜0.855の間に分布している。Rminの値が0.766以上となるtripletを検索することによって、/モチーフの候補を30本以下に絞ることが可能になる。 2)鎖の配列による8本の/モチーフの選出シミュレーション /バレル蛋白質内で、特に鎖がどのように並んで互いにパッキングして いるかを予測するために8本の鎖の配置を解析する。鎖を二次元に展開すると図2のような主鎖の水素結合様式が得られる。それぞれの鎖の最初のアミノ酸残基に対してC端側の鎖の最初の残基の位置は右側にシフトし、隣接する鎖間のシフト数Nを定めることができる。シフトの現われる鎖は/バレル蛋白質により異なり、70通りの可能性がある。1)で選出した/モチーフの候補から、「正しい8本のモチーフ」を「正しい水素結合様式」になるように選び出せれば三次元構造に組み上げることが可能になる。そのため、次の2種類の関数を組み合わせる。  図2./バレル蛋白質におけるシートの水素結合様式8本の鎖のN端側の残基を左側に示し、N端側の鎖からC端側の鎖へと順に上の行から下の行へと示す。第1行目にあるN端の鎖は第9行目にも示している。バレルの外側に向かう側鎖を持つアミノ酸を上側に、内側に向かう側鎖を持つものを下側に表示し、鎖間の水素結合を破線で示している。バレルの中心軸に対して鎖が円筒上で約36度傾いているため、鎖のシフトが現われる。上流の鎖の最初のアミノ酸残基に対して次の鎖のアミノ酸残基が同じ列から始まる場合にシフト数Nを0とし、その2残基分ずれる場合にNを1とし、4残基分ではNを2とする。いずれの/バレル蛋白質でもシフト数の総和は4となる。TIMの場合は、3、4、7、8番目の鎖のシフト数Nが1となり、他の鎖のNは0となる。 図2./バレル蛋白質におけるシートの水素結合様式8本の鎖のN端側の残基を左側に示し、N端側の鎖からC端側の鎖へと順に上の行から下の行へと示す。第1行目にあるN端の鎖は第9行目にも示している。バレルの外側に向かう側鎖を持つアミノ酸を上側に、内側に向かう側鎖を持つものを下側に表示し、鎖間の水素結合を破線で示している。バレルの中心軸に対して鎖が円筒上で約36度傾いているため、鎖のシフトが現われる。上流の鎖の最初のアミノ酸残基に対して次の鎖のアミノ酸残基が同じ列から始まる場合にシフト数Nを0とし、その2残基分ずれる場合にNを1とし、4残基分ではNを2とする。いずれの/バレル蛋白質でもシフト数の総和は4となる。TIMの場合は、3、4、7、8番目の鎖のシフト数Nが1となり、他の鎖のNは0となる。 a)シフト関数S:バレルにおいて、鎖のN端側のターン部分及び鎖は隣の鎖のターンや鎖と相互作用している。そのため、2つの鎖のアミノ酸残基の組み合わせによってシフト数Nが決まると考えられる。隣接する鎖間の正しいシフト数に対応する値が、他のシフト数に対する値よりも高くなるようなシフト関数Sを定める。  ここで、dsh(N,i,j’)は鎖m上の位置iと鎖m+1上の位置j’のアミノ酸残基の組み合わせの出現頻度値で、正しいシフト数Nに対するS.の値が最高になるように重みWsh(N,i,j’)を求める。この関数により、8本の/モチーフの候補に対して、一義的に/バレルを形成する可能性の有無及び可能な水素結合様式を判定できる。 b)パッキング関数P:/バレルの鎖は隣接する鎖だけでなく、2本先、3本先、4本先の下流側の鎖とも相互作用している。そこで、下流側の鎖との相関関係を考慮し、/バレル構造を実際に構成している/モチーフの組み合わせに対する値が他の組み合わせに対する値よりも高くなるようなパッキング関数Pを定める。  ここで、dp(k)(n",i,j")は鎖m上の位置iと鎖mに対してシフト数の和n"の位置にある鎖m+k上の位置j"のアミノ酸残基の組み合わせの出現頻度値で、真の構造に対するPの値が最高となるように、重みwp(k)(n",i,j")を求める。 結果・結論 表1に各蛋白質の選出シミュレーションの結果を示す。/バレル蛋白質、/蛋白質の1種であるRossmann foldでは、10〜30本程度の/モチーフ候補が検出されるのに対し、それ以外の蛋白質ではこのモチーフが8本未満しか検出されない場合が多い。  表1.選出シミュレーションの結果 表1.選出シミュレーションの結果 シフト関数Sで/バレル構造を形成する可能性があると判定される8本のモチーフの組み合わせは、/バレルでは複数検出されるのに対し、Rossmann foldではalcohol dehydrogenaseの1組を除いて検出されていない。さらに、パッキング関数Pで高得点を取る組み合わせはRossmann foldでは検出されないのに対して、バレル蛋白質では多数存在し、本物の8本の配列の組み合わせが最高得点となる。この結果は、この方法が一次構造によるバレル構造とそれ以外のドメイン構造の違いを識別することが可能であり、バレル構造として最良の構造を抽出可能であることを示す。/モチーフのような構造は、離れたポリペプチド鎖と相互作用することによって構造を構築している可能性が高く、離れたポリペプチド鎖との関係を考慮することによって立体構造を正しく予測することが可能になる。 抽出されたバレル構造を既知の同じ水素結合様式をとる三次元構造と比較して、相違するアミノ酸側鎖を置換し、エネルギー的に安定化させることにより三次元構造の構築も可能となる。今回開発した方法では/バレルのドメイン構造のみしか抽出できないが、Rossmann foldやImmuno-globulin foldなど他のドメイン構造の特性とその一次構造の統計データを求めることによって他のドメイン構造も抽出可能になることが期待される。 |
| 審査要旨 | | Triose phosphate isomerase(TIM)の樽状構造に代表される/バレルのドメイン構造は酵素全体数の10%近くに含まれると推定されている。/バレルでは,ヘリックスと鎖の対が/モチーフを成し,8本のモチーフの鎖が平行に配置してバレルを形成している。/バレル蛋白質は,アミノ酸配列の相同性から十数種のファミリーに分類されるが,ファミリー間の相同性は20%以下と低く,/バレルの起源と進化,およびその立体構造形成の機構の解明が課題となっている。 本研究では,アミノ酸配列から/バレル蛋白質の二次構造を予測し,/モチーフを立体的に配置するドメイン構造予測法の構築を目的とした。研究では,まず,三次元構造が既知のTIMやaldolaseなどを16種のファミリーに分類し,各々の蛋白質の一次構造と二次構造の相関を統計的に解析した。ついで,構造既知の20個の蛋白質について,統計的解析データに基づいて/モチーフを抽出し,モチーフの相対的な配置を二次構造の様式として得る4段階から成る手法を開発した。この手法で抽出したバレル構造を,同じ二次構造の様式をとる既知の三次元構造の蛋白質と置き換え,異なるアミノ酸の側鎖のモデリングと構造のエネルギー的な最適化を行えば,予測による三次元構造の構築に至る。 構造予測法の第1の段階は/モチーフの抽出である。アミノ酸20残基の領域で,/モチーフ中に出現する個々のアミノ酸残基(singlet)と,異なる位置のアミノ酸残基同士の2個の組み合わせ(doublet)の出現頻度を求めた。これら統計データをもとに,/モチーフを形成する傾向が強い配列を抽出するための/モチーフ関数を考案した。この段階では可能なモチーフの候補をできるだけ多数抽出することとしている。第2段階は/モチーフの候補の絞り込みであり,3残基のアミノ酸残基の組み合わせ(triplet)の寄与を,物理化学的性質に基づくアミノ酸残基同士の類似度として評価するtriplet消去法によっている。一次構造のデータベースから高い/モチーフの類似度を示すtripletを検索し,一定の上限値以下の類似度のtripletを含む領域を候補から除外するtriplet消去法を開発することにより,/モチーフの候補を小数に絞り込むことが可能となった。 第3段階は,隣接する鎖間の水素結合の整合性の評価であり,/モチーフの候補から可能な8本のモチーフの組み合わせを求める。/バレルの中心軸に対して鎖が円筒上で約36度傾いているため,水素結合が始まる鎖のアミノ酸残基の相対位置のシフトが現われ,いずれの/バレル蛋白質でもシフトの総和は8残基となっている。この関係を反映して隣接する鎖間のシフトを評価するシフト関数を考案することにより,8本のモチーフの候補に対して一義的に/バレルを形成する可能性の有無と可能な水素結合様式の判定を可能とした。さらに,隣接する鎖だけでなく,4本先までの下流側の鎖との配列上の相関関係を考慮しながら,/バレル構造を構成し易い/モチーフの組み合わせを評価するパッキング関数を考案した。この最終段階での評価によって,/モチーフの組み合わせとその立体的な配置を模式的に得ることが可能となった。 構造予測法を各種の蛋白質に適用した結果,/バレルとRossmann foldでは10〜30本程度の/モチーフの候補が抽出され,他の蛋白質では8本未満であった。シフト関数の適用により,/バレルを形成すると判定されるモチーフの組み合わせは,/バレルでは複数検出され,他の蛋白質ではRossmann foldのalcohol dehydrogenaseの1組のみであった。最終段階のパッキング関数で高値を示したものは/バレル蛋白質のみであり,本物の8本の組み合わせ配列を抽出できた。この結果は,本方法によればアミノ酸配列に基づいて/バレル蛋白質を識別し,適切な/バレル構造を抽出できることを示している。 このように本論文では,/バレル蛋白質の二次構造の詳細な統計的な解析,/バレルのドメイン構造構造予測法の開発,および,本方法により三次元構造が未知の蛋白質の一次構造データから/バレル構造が抽出可能であることを示している。本論文での/バレル構造の系統的な構造研究は,この構造の起源と進化の考察に基礎知見を与えるとともに,この構造を有する蛋白質の三次元構造の研究にも役立ち,酵素の構造化学,蛋白質構造生物学および薬学に寄与するところ大と評価した。以上により,本論文は博士(薬学)の学位の授与に値する内容を有すると認定した。 |