近年、長期にわたる血管内カテーテル留置などを原因とする院内感染症や消化管穿孔の合併症として、敗血症が重要視されている。敗血症はその半数がエンドトキシンショックに進行し、ショックの致死率は10%から時に90%と高率である。最近の研究により、エンドトキシンはマクロファージをはじめとする多種の細胞を刺激してインターロイキン1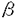 (interleukin 1、以下IL1)などの生体内活性物質を産生させ、これらを介してショックをおこすことが知られてきた。エンドトキシンショックにおいては全身血圧の低下がみられ、この原因としてIL1の血管平滑筋細胞への影響が重要視される。すなわち、IL1が血管平滑筋細胞において一酸化窒素(NO)合成酵素を誘導してNO/cyclic GMP(cGMP)産生系が活性化することによって血管弛緩がおこることが報告され、さらにはNO合成酵素(NOS)阻害薬のエンドトキシンショックへの臨床応用が報告されるに至った。しかし一方ではNOS阻害薬がすべてのエンドトキシンショックに有効というわけではなく、血管弛緩にNO以外の機構が関与している可能性がある。本研究はIL1の血管平滑筋に対する作用を明らかにすることを目的とした。 (interleukin 1、以下IL1)などの生体内活性物質を産生させ、これらを介してショックをおこすことが知られてきた。エンドトキシンショックにおいては全身血圧の低下がみられ、この原因としてIL1の血管平滑筋細胞への影響が重要視される。すなわち、IL1が血管平滑筋細胞において一酸化窒素(NO)合成酵素を誘導してNO/cyclic GMP(cGMP)産生系が活性化することによって血管弛緩がおこることが報告され、さらにはNO合成酵素(NOS)阻害薬のエンドトキシンショックへの臨床応用が報告されるに至った。しかし一方ではNOS阻害薬がすべてのエンドトキシンショックに有効というわけではなく、血管弛緩にNO以外の機構が関与している可能性がある。本研究はIL1の血管平滑筋に対する作用を明らかにすることを目的とした。 1)血管平滑筋におけるNOの合成促進とその脱感作 ラット大動脈標本を無菌的に採取し内皮を剥離した後、10%ウシ胎児血清を含むDulbecco’s modefied Eagle medium(DMEM)中で、37℃のCO2インキュベーター内で6および24時間、それぞれ器官培養したものを対照群とし、DMEM中にIL1(20ng/ml)を投与して同じく培養したものをIL1群とした。 6時間培養した対照群(対照-6時間群)ではphenylephrine(PHE)による最大収縮および収縮のEC50が一部抑制されたが、対照-24時間群ではそれ以上の抑制は見られなかった。この収縮抑制はNOS阻害薬のL-NMMA(100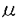 M)を収縮実験前に投与することで解除され、DMEM中で標本を培養することで少量のNOSが誘導されると考えられた。 M)を収縮実験前に投与することで解除され、DMEM中で標本を培養することで少量のNOSが誘導されると考えられた。 一方、IL1-6時間群ではPHEによる最大収縮が対照群の約61%まで抑制され、濃度作用曲線は対照群の約8倍右方に移動した。IL1-24時間群ではこの収縮抑制はさらに顕著になり、最大収縮は対照群の27%に減少し、濃度作用曲線も対照群の約38倍右方に移動した。またIL1-6時間群の組織内cGMP量は培養前の約27倍にまで大きく増加したが、IL1-24時間群では培養前の約8倍と減少した。 収縮実験時にL-NMMA(100M)の存在下でPHEを累積投与すると、IL1-6時間群では最大収縮、EC50ともほぼ対照群のレベルにまで回復した。しかしIL1-24時間群では対照群の約43%までしか最大収縮の回復は見られなかった。 以上の結果より、IL1を投与して6時間後にはNO合成の促進が起こるが、24時間後にはその脱感作が起こっている可能性が示唆された。 さらにIL1-24時間群に見られた収縮抑制はDMEM中に過剰のL-NMMA(1mM)を投与しても回復しなかった。また持続的にNOを放出するSNAP(10M)をDMEM中に投与して24時間培養した標本のPHE収縮は対照群と差がなかった。これらの結果より、IL1群において見られた収縮抑制効果は培養中に発生したNOに依存しないことが示唆された。 さらに、6時問および24時間培養をおこなった対照群およびIL1群の外来性のNOに対する反応性を検討した。対照群では培養6時間後、24時間後ともPHEによる収縮はsodium nitroprusside(SNP,1M)によりほぼ完全に抑制された。これに対してIL1群では6時間後ではSNPにより約47%の抑制しか見られず、24時間後には抑制効果は見られなかった。培養24時間後の組織内cGMP量を測定したところ、対照群ではSNP(1M)により静止状態の約33倍の組織内cGMP量の増加が見られたのに対し、IL1群では増加は2倍以下に過ぎなかった。これらの結果より、IL1で24時間処置することによりNOに対する反応の大部分が脱感作されている可能性が示唆された。さらにcGMPの膜透過性アナログである8-bromo-cGMP(100M)による収縮抑制に対照群とIL1群で差が見られなかったことから、グアニル酸シクラーゼの脱感作が起こっていると考えられた。 2) Interleukin 1によるNO非依存性の収縮抑制 以上の成績から、IL1を投与して24時間後にはNOに非依存性の収縮抑制機構が発現することが考えられたので、以下はこの機構について検討した。 細胞外からのCa2+流入のみに依存する高濃度K+収縮の[Ca2+]i-収縮張力関係について対照群、IL1群で比較したところ、差は見られなかった。また細胞膜に穴を開け細胞内のイオン環境を自由に操作できるようにしたラット腸管膜動脈スキンドファイバー標本の収縮はIL1により変化しなかった。これらの結果はIL1が収縮タンパク質を変化させないことを示している。 次にK+チャネルを阻害することによって膜電位を上昇させて収縮を起こすtetra-ethylammonium(TEA)による収縮について検討した。対照群ではTEAは濃度依存性に収縮を発生したが、IL1群ではTEAを30mMまで投与しても収縮は見られなかった。そこで次に静止膜電位の過分極を疑い、軽度の膜電位の上昇を引き起こして最大収縮の20%程度の収縮を起こす25.4mM KCl存在下でラット腸管膜動脈のIL1群にPHE又はTEAを投与したところ、収縮は対照群のレベルにまで回復した。また膜電位の測定結果からIL1によりTEAの脱分極作用が抑制されていることが示唆された。これらの結果はTEA感受性のK+チャネルをIL1が開口させることで収縮を抑制していることを示唆する。 次にIL1が作用するK+チャネルを特定するために各種選択的K+チャネル阻害薬を用いた実験を行った。ATP感受性K+チャネル開口薬であるlevcromakalim(1M)および阻害薬であるglibenclamide(1M)の投与により対照群のPHE収縮は変化したが、IL1群のそれは変化しなかった。またCa2+依存性K+チャネル阻害薬であるcharybdotoxin(0.1M)は対照群のPHE収縮を増強し、IL1群のそれもわずかに増強した。したがって、Ca2+依存性K+チャネルがIL1によって影響される可能性は低く、IL1によって開口されるK+チャネルはATP感受性K+チャネルや、TEAに感受性のある遅延整流K+チャネルである可能性が強い。 3) Interleukin 1による収縮抑制の機構 上述の収縮抑制がどのような細胞内情報伝達系によって発現されるかを検討するために、24時間の培養中にIL1と各種薬物を共存させて、その後の収縮反応を調べた。 タンパク質合成阻害薬であるcycloheximide(10M)をIL1と共に投与することによりPHE収縮は最大収縮、EC50ともに対照群のレベルまで回復し、IL1の収縮抑制効果の発現にタンパク質合成が関与していることが強く示唆された。 セリン/スレオニンキナーゼの非選択的阻害薬であるH-7(10M)は、対照群およびIL1群のPHE収縮になんら効果を持たず、セリン/スレオニンキナーゼはIL1の収縮抑制効果の発現に関与していないことが示唆された。 またgenistein(100M)、tyrphostin(1M)の2種のチロシンキナーゼ阻害薬が対照群およびIL1群のPHE収縮になんら効果を持たない一方、herbimycin-A(1M)はIL1群のPHEによる最大収縮を対照群のそれ近くまで回復させた。これらのチロシンキナーゼの阻害薬はそれぞれ性格が異なると考えられるため、IL1の情報伝達系にかかわるチロシンキナーゼはherbimycin-Aのみに感受性を持つものであることが示唆される。しかし、PI3キナーゼを阻害するwortmannin(0.1M)がIL1の収縮抑制作用に影響を与えなかったことから、PI3キナーゼがIL1の情報伝達系に含まれている可能性は薄い。 さらに非ステロイド性抗炎症薬であるaspirin(3mM)によってIL1による収縮抑制効果は一部解除されたが、同じく非ステロイド性抗炎症薬であるindomethacin(10M)はこのような作用を持たず、IL1の情報伝達系にはaspirinに特異的に反応する経路が存在することが示唆された。 4)まとめ 本研究において、IL1によりNO産生系の活性化とこれに続く脱感作がおこることが示唆された。しかしNO産生系の脱感作後もIL1による強い収縮抑制がみられ、NOに依存しない収縮抑制機構が発現していることが考えられた。この抑制機構は、K+チャネルの開口による細胞膜の過分極による可能性が高い。また、この抑制機構はタンパク質合成を介して発現し、チロシンキナーゼに依存していることが明らかとなった。 |  に関する研究
に関する研究