本研究は肥大型心筋症(HCM)患者の心筋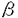 ミオシン重鎖(cMHC)遺伝子変異スクリーニングを行なうとともに、同定したミオシン重鎖変異の収縮機能への影響を解析を試みたものであり、下記の結果を得ている。 ミオシン重鎖(cMHC)遺伝子変異スクリーニングを行なうとともに、同定したミオシン重鎖変異の収縮機能への影響を解析を試みたものであり、下記の結果を得ている。 1.75名のHCM患者のよりゲノムDNAを抽出し、cMHC遺伝子変異スクリーニングを、従来用いられてきたsingle-strand conformation polymor-phism(SSCP)法と今回初めてcMHC遺伝子変異スクリーニングに応用されたOrphan Peak Analysis(OPA)法で行い、両方の方法で1名の女性患者にミスセンス変異(731Pro→Leu)を同定した。家族歴および遺伝子解析から、母方の祖母由来の家族性HCMと推定された。cMHCコドン731の変異はこれまで報告がない新しいミスセンス変異である。 2.cMHCコドン731はアクチン結合部位の比較的近傍にあり、変異によるアクチン・ミオシン相互作用への影響をpeptide competition法で解析した。その結果、変異ミオシンは正常ミオシンと比較し、より強力にアクチンと相互作用することが示唆された。 3.ミオシンのアミノ酸の電荷の変化を伴わない変異は予後が良好であると報告されているが、本家系では予後不良であり、予後を決定するのは電荷の変化よりむしろ変異によるミオシンの構造、機能への影響であると示唆された。 4.75名のHCM患者(2名家族性、73名孤発性)のcMHC遺伝子のスクリーニングの結果、家族性患者2名中1名(50%)に変異が同定され、73名の孤発性患者に変異は発見されなかった。孤発性HCMでは、cMHC遺伝子変異の頻度が低いことが示された。 5.OPA法は技術的に複雑であるが、本研究で示したとおりに実験操作を行なうことにより多数のサンプルの遺伝子変異スクリーニングを高感度に行うことが可能であることを示した。 以上、本論文は、OPA法を遺伝子点変異スクリーニング法として実用化し、HCM患者に新たなcMHC変異を同定するとともに、peptide competition法を用いて今回発見した変異の収縮機能への影響を明らかにしたものであり、学位の授与に値するものと考えられる。 | 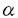 -トロポミオシン遺伝子にもそれぞれ点変異が報告されている。c
-トロポミオシン遺伝子にもそれぞれ点変異が報告されている。c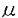 Mのペプチド20-Nまたは20-M)を含む溶液中で測定した。ペプチド20-Nはc
Mのペプチド20-Nまたは20-M)を含む溶液中で測定した。ペプチド20-Nはc