| 内容要旨 | | リチウムジイソプロピルアミド(LDA)に代表される単配座型リチウムアミドは、求核性の弱い強塩基としてリチウムエノラートの生成に広く用いられる。このカルボニル化合物の脱プロトン化反応は、リチウムアミドのmonomerがカルボニル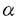 位のプロトンを引き抜くIrelandらの6員環遷移状態モデルを仮定して考えられている(図1)。1)しかし、近年、Collumらによりリチウムアミドが溶液中様々な会合状態で存在することが明らかにされ、8員環等の遷移状態モデルの可能性も示唆されている。2) 位のプロトンを引き抜くIrelandらの6員環遷移状態モデルを仮定して考えられている(図1)。1)しかし、近年、Collumらによりリチウムアミドが溶液中様々な会合状態で存在することが明らかにされ、8員環等の遷移状態モデルの可能性も示唆されている。2) 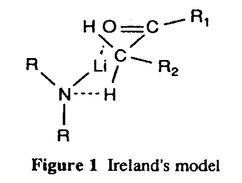 Figure 1 Ireland’s model Figure 1 Ireland’s model 先に、我々及びSimpkinsらは単配座型キラルリチウムアミド(1)を用いた4-tert-butylcyclohexanone(2)の不斉脱プロトン化反応において、LiCl添加による選択性の向上について報告した。3)すなわち、リチウムアミドにエノラート捕捉剤であるTMSClをあらかじめ加えてから脱プロトン化反応を行なうCoreyのinternal quench法4)では90%eeと非常に高い選択性で対応するシリルエノールエーテル(3)を与えたのに対し、脱プロトン化後TMSClを加えるexternal quench法では44%eeと選択性が著しく低下した。この差異は系内で生じるLiClに起因するのではないかと考え、external quench法において予めLiClを系内に加え反応を行ったところ選択性が88%eeへと大きく向上した(表1)。 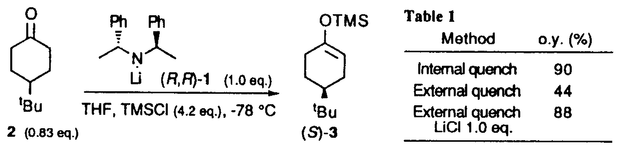 Table 1 Table 1 以上の結果をふまえ、我々は種々の条件でのリチウムアミドの会合状態をNMRにより捉え、反応の選択性と相関づけることにより、脱プロトン化反応の機構解明へのアプローチを試みた。 1.Li halides添加によるリチウムアミド(1)の溶液構造の変化と選択性との相関 リチウムアミド1を用い、LiCl、LiBr、LiI共存下での脱プロトン化反応を行った。-78℃では、LiCl添加時(0.5,1.0,3.0当量)には87〜88%eeを示したのに対し、LiBrでは1.0当量を加えると63%ee、3.0当量を加えると86%eeを示した。LiIの添加では選択性が向上しなかった(表2)。このリチウム塩による選択性の差異は、ハライドにより会合状態が異なることに由来するのではないかと考え、[6Li,15N]-(S,S)-1を合成し、6LiCl、6LiBr、6LiI添加によるTHF中-115℃での1の溶液構造の変化を6Li,15N-NMRにより捉えた。その結果、LiCl非共存下では1はmonomer Aとdimer Bの状態で存在した(図2(a))。0.5,1.0当量のLiClを加えると、1とLiClのmixed aggregates C、D(図2(b),(c))が生成し、3.0当量のLiCl共存下では単一の会合種Dへと収束することがわかった(図2(d))。また、LiBr3.0当量の添加ではmixed aggregate Eの形成が認められるものの、依然としてA、Bが存在した(図2(e))。LiI共存下ではA、Bのみ捉えられた(図2(f))。 NMR測定に対応した-114℃で脱プロトン化反応を行い、上記会合状態とそれらが与える反応の選択性との相関を精査した(表2)。A、Bのみ存在する図2(a)の条件では41%eeと低い選択性であるのに対し、C、Dが十分に存在する図2(c)の条件では91%eeと高い選択性を示した。C、Dの寄与の大小は不明だが、Dのみ存在する図2(d)の条件で91%eeと高いeeを示すことから、少なくともDは高選択性を与える会合種であることがわかった。また、LiBr3.0当量の添加では71%eeであるが、-78℃では86%eeで3を与えており、Eもまた高選択性を与える会合種であると考えられる。LiIは反応の選択性に影響を与えないが、それは会合状態に関与しないためであると判明した。LiCl0.5当量、LiBr1.0,3.0当量添加時には-114℃での選択性は、-78℃での選択性より低下した。-114℃では会合種間の平衡が遅いため、mixed aggregatesの存在比が低い初期の会合状態を反映してmonomer、dimerからの反応が進行しやすいのに対し、-78℃では会合種間の平衡が速いため、より反応活性が高くかつ高選択性を与えるmixed aggregatesから反応する傾向が強まり選択性が向上したと考えられる。反応系内にはリチウムエノラートが存在するが、リチウムアミドの会合状態には影響を与えないことをNMRにより確認している。 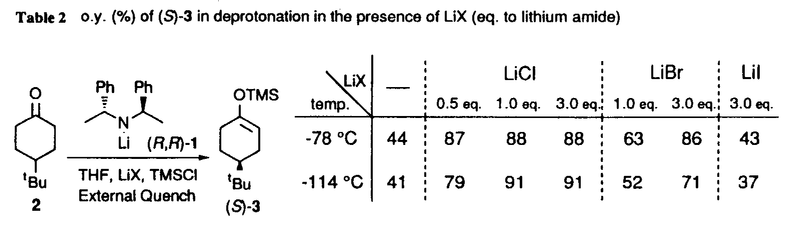 Table 2 o.y.(%)of(S)-3 in deprotonation in the presence of LiX(eq.to lithium amide) Table 2 o.y.(%)of(S)-3 in deprotonation in the presence of LiX(eq.to lithium amide)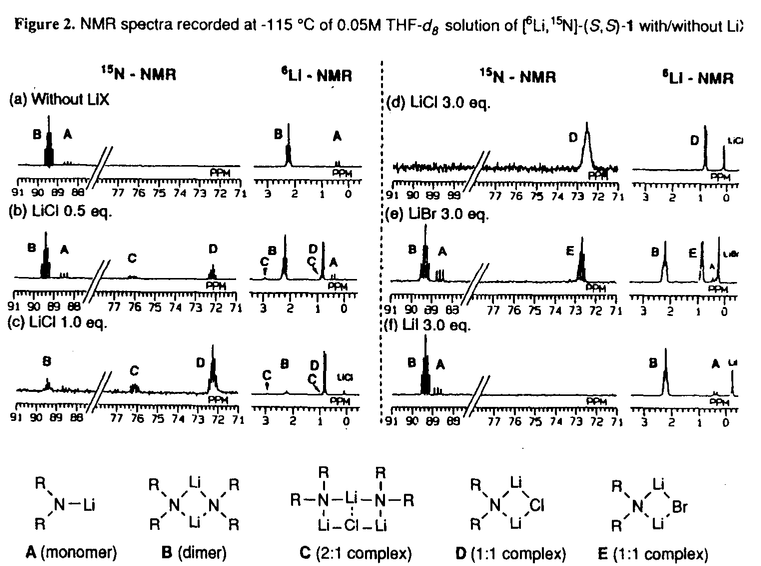 Figure 2.NMR spectra recorded at-115℃ of 0.05M THF-d8 solution of[6Li,15N]-(S,S)-1 with/without Li)2.Monomerとmixed aggregateの比較 Figure 2.NMR spectra recorded at-115℃ of 0.05M THF-d8 solution of[6Li,15N]-(S,S)-1 with/without Li)2.Monomerとmixed aggregateの比較 高選択性を与えるリチウムアミド/LiX mixed aggregatesから反応が進行する際、Irelandモデルのようにmonomerを経由するか否か検証するため、以下の実験を行った。 2.1DME中での溶液構造と反応の選択性 DME中、1はmonomer F、LiClを添加するとmixed aggregate Gとして存在する。不斉脱プロトン化反応の結果と比較するとFからは61%ee、Gからは88%eeで生成物を与える(表3)。これらの結果から、mixed aggregateはmonomerよりも高選択性を与えること、遷移状態にLiClが関与することが判明した。 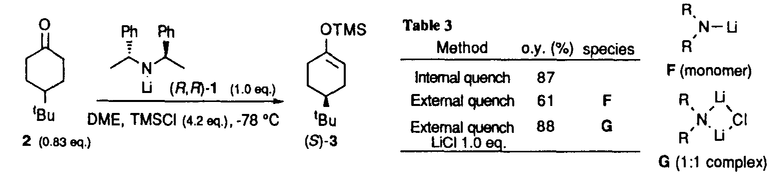 Table 32.2二座配位型リチウムアミド(4) Table 32.2二座配位型リチウムアミド(4) 当教室で開発された二座配位型キラルリチウムアミド(4)は分子内キレートにより固定された不斉場を持ち、不斉脱プロトン化反応で高い選択性を示す。5)4はTHF中においてmonomerHとして存在するが、LiClの添加によりHからmixed aggregate Iへと会合状態が変化し、それに伴い反応の選択性の向上が認められた(表4)。反応はmonomerではなくmixed aggregateを経由して進行すると考えられる。 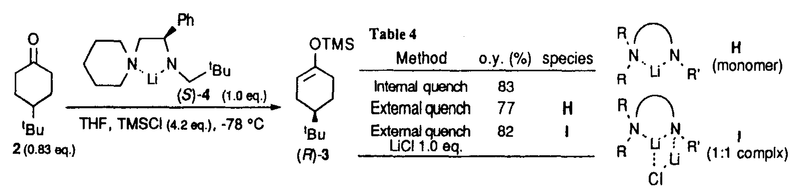 Table 43.リチウムアミドとシクロヘキサノン4位置換基との立体効果 Table 43.リチウムアミドとシクロヘキサノン4位置換基との立体効果 不斉脱プロトン化反応の遷移状態でのリチウムアミドの立体配置を考察するために以下の実験を行った。フェニル基上に嵩高い置換基(3,5-di-tert-butyl)を導入したリチウムアミドを用いると、2を基質とした場合、90%ee(S)から16%ee(R)へと選択性の向きが逆転したのに対し、5を基質とすると、90%ee(S)から68%ee(S)と選択性の若干の低下のみ認められた(表5)。これらの結果より、遷移状態においてリチウムアミドのフェニル基(R2)とcyclohexanone 4位の置換基(R1)とは互いに近傍に位置していると考えられる。 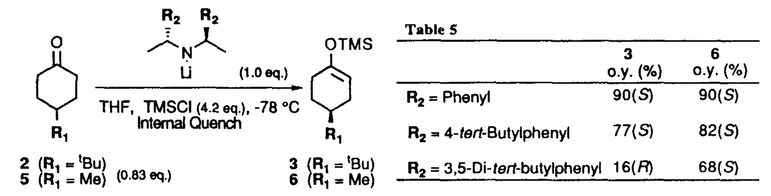 Table 54.まとめ Table 54.まとめ 以上の結果より、internal quench法を用いた脱プロトン化反応は、monomerではなくリチウムアミド/LiX mixed aggregatesを経由して進行すると考えられる。Collumらの結果2)も考慮すると、本不斉反応は、六員環ではなく右図のような8員環の遷移状態が考えられる。 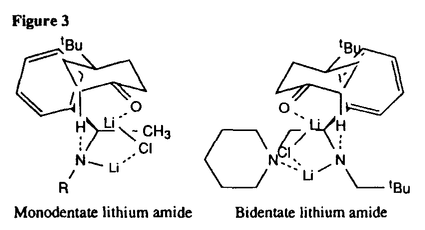 Figure 3文献1)Ireland,R.E.;Mueller,R.H.;Willard,A.K.J.Am.Chem.Soc.1976,98,2868.2)(a)Galiano-Roth,A.S.;Kim,Y-J.;Gilchrist,J.H.;Harrison,A.T.;Fuller,D.J.;Collum,D.B.J.Am.Chem.Soc.1991,113,5053;(b)Romesberg,F.E.;Collum,D.B.J.Am.Chem.Soc.1995,117,2166.3)(a)菅沢 形造、野口 博司、古賀 憲司 日本薬学会第113年会(1993)(b)Bunn,B.J.;Simpkins,N.S.J.Org.Chem.1993,58,533.4)Corey,E.J.;Gross,A.W.Tetrahedron Lett.1984,25,495.5)Sato,D.;Kawasaki,H.;Shimada,I.;Arata,Y.;Okamura,K.;Date.T.;Koga,K.J.Am.Chem.Soc.1992,114,761. Figure 3文献1)Ireland,R.E.;Mueller,R.H.;Willard,A.K.J.Am.Chem.Soc.1976,98,2868.2)(a)Galiano-Roth,A.S.;Kim,Y-J.;Gilchrist,J.H.;Harrison,A.T.;Fuller,D.J.;Collum,D.B.J.Am.Chem.Soc.1991,113,5053;(b)Romesberg,F.E.;Collum,D.B.J.Am.Chem.Soc.1995,117,2166.3)(a)菅沢 形造、野口 博司、古賀 憲司 日本薬学会第113年会(1993)(b)Bunn,B.J.;Simpkins,N.S.J.Org.Chem.1993,58,533.4)Corey,E.J.;Gross,A.W.Tetrahedron Lett.1984,25,495.5)Sato,D.;Kawasaki,H.;Shimada,I.;Arata,Y.;Okamura,K.;Date.T.;Koga,K.J.Am.Chem.Soc.1992,114,761. |
| 審査要旨 | | リチウムジイソプロピルアミド(LDA)は代表的な単配座型リチウムアミドであり、求核性の弱い強塩基として、カルボニル化合物の脱プロトン化によるリチウムエノラートの合成に広く用いられている。単配座型の光学活性リチウムアミド(1)を用いてプロキラルな4-tert-butylcyclohexanone(2)を脱プロトン化し、対応するシリルエノールエーテル(3)として単離するとき、不斉脱反応が起こることは既に知られていた。本不斉反応を詳細に検討した結果、リチウムエノラートの捕捉剤である塩化トリメチルシリル(TMSCl)をあらかじめ加えておく(internal quench法)と高い不斉誘起が見られるが、脱プロトン化後にTMSClを加える(external quench法)と不斉誘起の程度は著しく低下し、external quench法においてあらかじめLiClを加えておくと、不斉誘起の程度が回復することを見出した。本研究は、上記の現象を解明するための検討を行い、本脱プロトン化反応について新しい機構を提唱したものである。 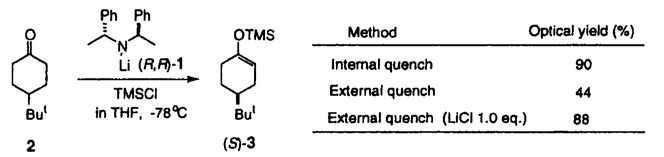 図表 図表 (R,R)-1を用いた2の脱プロトン化反応をTHF中external quench法で行い、これに最初からLiCl、LiBr、あるいはLiIを添加したときの3の光学純度を調べた。このリチウム塩による顕著な不斉収率への影響は、1の溶液中での会合状態の違いによるものではないかと推定した。 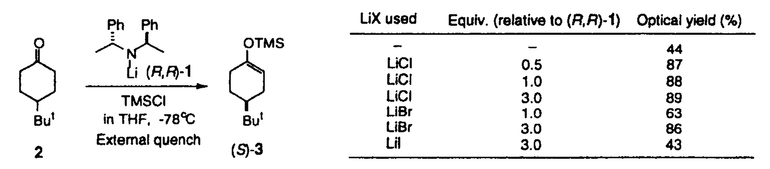 図表 図表 そこで、[6Li,15N]-(S,S)-1を合成し、そのTHF-d8溶液中での会合状態が、6LiCl、6LiBr、6LiIによってどのように変化するかを6Li-および15N-NMRによって調べた。その結果、光学活性1はリチウム塩を添加しないときは僅かのAを含むBとして存在すること、LiClを添加するとC、D(X=Cl)が現れ、3当量のLiClの存在ではD(X=Cl)が最も多くなること、LiBr添加でも同様の傾向があるが、3当量のLiBrの存在でもD(X=Br)の他にほぼ同量のBと少量のAが存在すること、LiIの添加は無添加の場合と変化がないこと、等が明らかとなった。この結果は、リチウム塩の添加による本不斉脱プロトン化反応の立体選択性の変化とよく符号している。 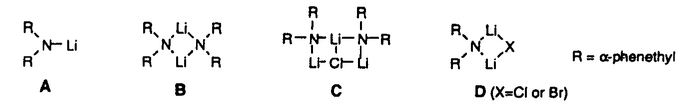 図表 図表 次に、本不斉脱プロトン化反応の遷移状態での反応基質とキラルリチウムアミドとの間の立体的相互作用を調べるため、2および4-methylcyclohexanone(4)を反応基質とし、(R,R)-1およびそのフェニル基上に嵩高い置換基を導入したキラルリチウムアミド((R,R)-5、(R,R)-6)を用いたときの反応を行った。その結果から、本反応の遷移状態では、基質であるシクロヘキサノンの4位の置換基とリチウムアミドのフェニル基上の3位および5位の嵩高い置換基とは互いに近傍に位置していると結論した。 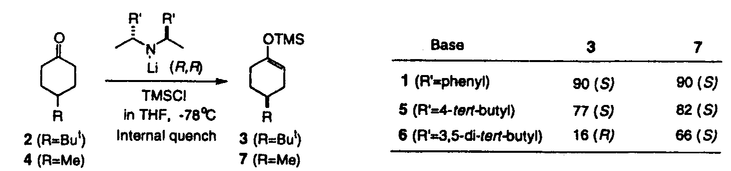 図表 図表 これらの結果を総合し、internal quench法を用いた脱プロトン化反応は、リチウムアミドがDの会合状態で反応し、従来考えられていた6員環遷移状態ではなく、8員環遷移状態を経由するものと提唱した。 以上、本研究は、単配座型リチウムアミドの溶液構造を明らかにすることによて、これらの塩基を用いたカルボニル化合物の脱プロトン化反応の反応機構に新しい考えを導入したもので、有機合成化学の進歩に寄与するものであり、博士(薬学)の学位に値するものであると認める。 |