本研究は、代表的なG蛋白質共役受容体であるムスカリン性アセチルコリン受容体の高次構造の解明を最終目標として、構造解析に適した種々の変異体を作成してバキュロウイルス-Sf9系を用いて発現し、それらの生化学的性質を調べたものであり、以下の結果を得ている。 まず、構造多様性排除や結晶性の向上、発現・精製効率の改善を目的としてムスカリン性アセチルコリン受容体m2サブタイプの各種変異体を構築し、バキュロウイルス-Sf9系で発現させた。作成した変異体はいずれもリガンド結合活性を保持していた。第三細胞内ループを欠失した変異体は発現量が1.2-1.5倍に向上しており、また細胞内プロテアーゼによる分解を受けにくかった。糖鎖付加部位の除去、シグナルペプチドとFLAGエピトープの融合、ヒスチジンタグの融合によっては野生型と比較して発現量は変わらなかった。Mycエピトープの融合、ポリヘドリン蛋白質N末11残基の融合、パルミチル化部位の除去によっては発現量が3/4-1/2に低下した。糖鎖付加部位の除去、ヒスチジンタグの融合、パルミチル化部位の除去を行った変異体については更に詳細な生化学的解析が行なわれた。 第一に、糖鎖の付加による構造多様性の排除を目的として、N末細胞外ドメインのN-グリコシル化のコンセンサス配列を満たすアスパラギン残基をアスパラギン酸に変換した変異体が作成され、その活性が調べられた。リガンドアフィニティーゲルABT(aminobenztropine)アガロースにより精製したm2受容体標品をウシ肺より精製したG蛋白質Gi2とともにリン脂質膜に再構成し、アゴニスト刺激によるG蛋白質への[35S]GTPgS結合促進活性、およびグアニンヌクレオチド非存在下での受容体のアゴニスト高親和性結合を調べたところ、野生型受容体と変わらないことが確認された。 第二に、C末端細胞内ドメインのパルミチル化について調べられた。Sf9細胞を[3H]パルミチン酸中で培養すると、野生型受容体は標識されたが、457番目のシステイン残基をアラニンに変換した変異体は標識されず、Cys457がm2受容体のパルミチル化部位であることが示された。パルミチン酸による標識は、Sf9細胞をアゴニスト存在下で培養することにより促進された。精製標品による再構成実験で、変異体もアゴニスト存在下でG蛋白質への[35S]GTP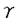 S結合を促進したが、その速度は野生型の約1/2に低下していた。G蛋白共役受容体キナーゼによるリン酸化には野生型と変異体で変化がないことが示された。 S結合を促進したが、その速度は野生型の約1/2に低下していた。G蛋白共役受容体キナーゼによるリン酸化には野生型と変異体で変化がないことが示された。 第三に、リガンドゲルに代わる効率よい精製法を求めて、C末端にヒスチジン6残基を融合した受容体を発現させ、その受容体を金属キレートゲルにより精製する方法を検討した。種々の金属イオンを結合させたChelating Sepharoseを用いて、ヒスチジン6残基を融合した受容体の精製効率を比較した。溶出にはイミダゾールを用いた。コバルトイオン(Co2+)を結合させたゲルを用いた場合最も精製効率が良かった。再構成実験により、ヒスチジン6残基のC末端への融合が受容体とG蛋白質との相互作用に影響を及ぼさないこと、Co2+ゲルを用いて精製した受容体もG蛋白質を活性化できることが確認された。ヒスチジンタグとCo2+ゲルを用いる方法はリガンドゲルによる精製法が確立されていない受容体に応用できる。また収率がよく受容体を安定に取り扱うことができるので、高次構造解析に用いる標品の調製に適している。 以上、本論文はムスカリン受容体の種々の変異体を作成し、バキュロウイルス-Sf9系で発現、精製し、その性質を生化学的に解析したものである。G蛋白質共役型受容体の多くは組織中の存在量が少なく、効率の良い精製方法が確立しているものもわずかであるため、その高次構造の研究はほとんど進んでいない。本研究はG蛋白質共役型受容体の高次構造解析に有用な種々の変異体を作成してその性質を明らかにし、また精製法を開発したもので、学位の授与に値するものと認められる。なお、本研究で述べた結果は芳賀達也との共同研究として公表されているが、殆ど全ての実験は論文提出者によって行われ、結果の解釈・考察の主要部分も論文提出者によって行われた。 |  アドレナリン受容体はC末端がパルミチル化を受けることが知られている。m2受容体についてもC末端細胞内ドメインのCys457残基がパルミチル化を受けると推定されていた。組み替え体ウイルスを感染させたSf9細胞を[3H]パルミチン酸中で培養したところ、野生型受容体は標識されたが、Cys457をアラニンに変換した変異体は標識されなかった。この結果は、Cys457がm2受容体のパルミチル化部位であることを示す。このパルミチン酸による受容体の標識は、Sf9細胞のアゴニスト刺激により促進された。この脂質修飾の受容体の機能に及ぼす役割を更に調べる目的で、パルミチル化部位の変異体を精製し、その性質をG蛋白質との再構成系で調べた。変異体もアゴニスト存在下でG蛋白質への[35S]GTP
アドレナリン受容体はC末端がパルミチル化を受けることが知られている。m2受容体についてもC末端細胞内ドメインのCys457残基がパルミチル化を受けると推定されていた。組み替え体ウイルスを感染させたSf9細胞を[3H]パルミチン酸中で培養したところ、野生型受容体は標識されたが、Cys457をアラニンに変換した変異体は標識されなかった。この結果は、Cys457がm2受容体のパルミチル化部位であることを示す。このパルミチン酸による受容体の標識は、Sf9細胞のアゴニスト刺激により促進された。この脂質修飾の受容体の機能に及ぼす役割を更に調べる目的で、パルミチル化部位の変異体を精製し、その性質をG蛋白質との再構成系で調べた。変異体もアゴニスト存在下でG蛋白質への[35S]GTP