本研究は、癌化シグナル伝達機構を明らかにするために、ラット線維芽細胞(NRK49F)の変異株を樹立し、どのような因子が癌化シグナル伝達に関わっているのかを解析したものであり、下記の結果を得ている。 1.EGF及び、TGF-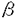 の存在下で可逆的に癌化を起こすNRK49Fを変異原処理し、独立した2回のスクリーニングからそれぞれ1つずつ計2つの癌化を起こさない劣性変異株456及び、変異株111を樹立した。 の存在下で可逆的に癌化を起こすNRK49Fを変異原処理し、独立した2回のスクリーニングからそれぞれ1つずつ計2つの癌化を起こさない劣性変異株456及び、変異株111を樹立した。 2.変異株456に癌遺伝子を導入し、その感受性を検討したところ、EGF受容体に近いv-erbB遺伝子には感受性を示さず、活性化型c-erbB2には非常に低い感受性を示した。またPDGF受容体に近いv-fms、Ki-ras,v-src,v-mos,ポリオーマミドルT抗原などによる癌化シグナルには、正常に癌化形質を示した。これらより、変異株456の変異点はEGF受容体の下流で、rasよりも上流であることが示唆された。 3.この変異株456の感受性はすでに樹立解析がなされているアダプター分子crkII遺伝子に変異のある変異株23と同一のものである。しかしながら、変異株23と456の融合細胞を作成し、解析した結果、互いに変異を相補する事がわかり、これらは異なった遺伝子に変異があることが示唆された。さらに、アダプター分子近傍で機能していると考えられる遺伝子、crkI、grb2、shc、sos、C3Gといった因子による相補活性を検討したが、相補する因子は存在しなかった。これらから、変異株456の変異因子はアダプター分子近傍で、未知の因子であると考えられた。 4.変異株111に癌遺伝子を導入し、その感受性を検討したところ、用いたすべての癌遺伝子について非感受性であった。特にEGFシグナルの最下流に存在する癌遺伝子の一つであるv-rafについて、その蛋白発現量と癌化能について詳細に検討したところ、rafの下流に変異点が存在していることが示唆された。 5.変異株111の対数増殖期の増殖特性は親株と変わらないが、一旦G1期に停止させ、その後細胞周期を再スタートさせると、親株に比べ、S期の開始が著しい遅延が観察された。実際親株が10時間程度でS期を開始するのに比べ、変異株111は20時間を要した。また、S期初期に停止後の再スタートに関しては問題ないことから、この変異株ではG1期の再スタートに何らかの以上が生じていることが示唆された。 6.変異株111を足場のない状態でG1期に停止後、癌化シグナルで再スタートさせ、G1期の進行のどこに異常が生じてS期の開始ができないのかを明らかにするために、G1期を制御している因子群の発現を経時的にRNA及び蛋白質レベルで解析した。変異株111は足場のない状態ではS期に進行できないが、そのときにG1期初期を制御しているサイクリンD1の発現の誘導が著しく減少していた。またそれに続くサイクリンEの発現誘導も起きていなかった。このことより、この変異株がS期に進行しないのはサイクリンD1の誘導の阻害が一原因になっていることが示唆された。 以上、本論文はラット線維芽細胞NRK49Fの変異株の解析から、アダプター分子近傍の癌化シグナルを伝える新たな未知の因子の存在の可能性と、通常の足場依存的増殖開始メカニズムと、癌化に特異的な足場非依存的増殖との接点にサイクリンD1分子が深く関わっている可能性を示唆した。本研究はこれまで未知に等しかった癌化シグナル伝達の細胞レベルでの解明に重要な貢献をなすと考えられ、学位の授与に値するものと考えられる。 |