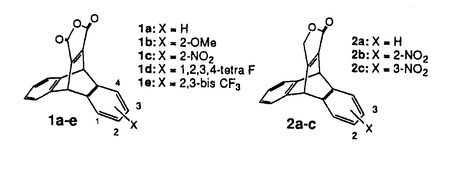学位論文要旨
| No | 112869 | |
| 著者(漢字) | 岡本,巌 | |
| 著者(英字) | ||
| 著者(カナ) | オカモト,イワオ | |
| 標題(和) | π-π軌道相互作用によるπ面非対称化と反応面選択 | |
| 標題(洋) | ||
| 報告番号 | 112869 | |
| 報告番号 | 甲12869 | |
| 学位授与日 | 1997.03.28 | |
| 学位種別 | 課程博士 | |
| 学位種類 | 博士(薬学) | |
| 学位記番号 | 博薬第780号 | |
| 研究科 | 薬学系研究科 | |
| 専攻 | ||
| 論文審査委員 | ||
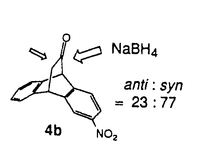
| 内容要旨 | オレフィンやケトンの作るπ面における反応面選択性は多数研究されており、多くは立体効果によって決定されている。そして立体の大きさや立体電子効果に基づくCram則やFelkin-Anhモデルが提出されてきた。本研究は、立体効果によらない場合でもπ軌道の非対称化によって反応面を決定しうるという考えに基づき、ジベンゾビシクロ[2.2.2]オクタトリエン骨格を有するマレイン酸誘導体(1)、フラノン誘導体(2)をデザイン・合成し、そのπ面選択特性を調べることを目的とした。1、2の一方のベンゼン環に置換基を導入すると、反応中心のπ面は立体効果ではなくπ-π軌道相互作用により非対称化する。 ジエノファイル1a-eに対し、1,3-butadiene(BD)、及び2,3-dimethyl-1,3-butadiene(DMBD)の直鎖状ジエンとのDiels-Alder反応を行った。これらの反応においては、anti及びsynの2種の生成物が得られる。無置換体1aは面対称な基質であるので当然その生成比に偏りはない。しかし置換基を導入した基質においては反応面に選択性が発現した。そしてその生成比(Table1)には、2種のジエンとの反応に共通の傾向が見られた。メトキシ体1bでは偏りはなく、電子吸引性の置換基(NO2,tetraF,bisCF3)を導入すると、anti付加がsyn付加よりも多く進行し、その偏りはbisCF3の場合に76:24になった。  次に、環状ジエンであるシクロペンタジエンとの反応を行った。この場合には面選択性とendd exo由来の4種の生成物がある(Table2)。endo付加における面選択性は、直鎖状ジエンの場合と同様の傾向がみられた。つまり、メトキシ体1bでは偏りはないが、電子吸引性の置換基を持つ1c-eでは、anti付加が優先する。exo付加においてもこの傾向は同様であるが、その比はendo付加及び直鎖状ジエンの場合に比べてやや小さくなっている。これらのDiels-Alder付加体は単離後に反応条件に戻しても異性化が見られないことから、π面選択は速度論的に決定されていると考えられる。  ジエノファイル1b-eにおける付加反応の置換基による反応速度変化を評価するために、1aと1b-eとの1:1混合とDMBDとのDiels-Alder反応を行い、その二次反応速度定数の相対値を測定した(Table3)。メトキシ基は反応に影響を及ぼさないことは明らかである。一方、電子吸引性の置換基は、anti付加において反応を加速しているが、syn付加においては、この加速は見られない。また、tetraF体(1d)においてはanti、synともに大きな加速がみられる。  a,β-不飽和ラクトン2a-cに対しては、まず塩基触媒下でのチオールの求核的1,4-共役付加反応を行った。この基質はラクトン構造のためtrans付加は進行せずにcis付加のみが起きる。この不飽和ラクトンにおいてもニトロ基の導入により面選択が出現し、ニトロ基の位置の異なる2b,2cともにsyn付加がanti付加よりも優先した(Table4)。付加生成物を単離後、反応条件に戻しても異性化はほとんど起こらないことから、ここでも反応面選択は速度論的に決定されていると考えられる。高極性溶媒を用いるとsyn面選択性は増大しているが、syn付加がanti付加よりも優先するという傾向は変わらないものである。 次にクラウンエーテル存在下でのシアン化カリウムの共役付加反応を行った。この場合にも生成物はantiとsynとの2種が得られる。この反応においてもニトロ体は2b,2cともにsyn付加がanti付加よりも優先して進行している(Table5)。反応面選択性はチオールの反応と同様である。   電子吸引性置換基を導入すると、1のDiels-Alder反応においてはanti付加が優先して進行したのに対し、2の求核的共役付加反応においてはsyn付加優先であった。この電子吸引性置換基によるsyn面選択性は、同様の骨格を有するケトン4bに対するNaBH4の求核反応においても観測された。 以上の反応の面選択は、反応基質分子のπ軌道の非対称化に起因すると考えられる。その非対称化は、分子全体を3個の部分構造(π反応中心と2つのアロマティック部分)に分割し、各部分構造のπ軌道から基質分子のπ軌道を作ると考えた場合、その軌道相互作用の大きさの違いにより引き起こされる(Scheme 1)。  また、ジエノファイル1、ラクトン2及びケトン4においては共にそのLUMOが反応に関与していると考えられる。分子のLUMOは、各部分構造のπ*軌道が同位相で相互作用して作られると考えられるが、アロマティック部分は共通であり、電子吸引性置換基をもつ1c-e、2b-cおよび4bともに反応中心及び置換基のあるベンゼン環に大きなローブがある(Scheme2)。これらの反応が異なるπ面選択性を示したことは注目すべきである。 求核的反応では、この同位相の広がりがチオエトキサイドまたはシアナイド(HOMO)の攻撃に有利である(Scheme2B)。これはケトン4に対するハイドライドの攻撃においても同様である。非プロトン性高極性溶媒における選択性の増大は、求核種であるフリーのチオエトキサイド濃度が増加し、その相互作用がより重要となるためと考えられる。  Diels-Alder反応において、NO2およびbisCF3の導入による反応の加速がsyn付加では観測されていない。また、1c-eのシクロペンタジエンとの反応でのendo/exo比がsyn付加では無置換体1aとは大きく異なる比を持っている。これらのことから、syn付加の場合に反応を妨げる相互作用が存在することが示唆される。Diels-Alder反応において反応するのはジエンのπ軌道である。π軌道の反対側のロープとジエノファイルのアロマティックπ*軌道とは逆位相となり軌道非相互作用系となってしまい、syn付加は不利となる。このため反応はanti優先で進行すると考えられる(Scheme2A)。また同時に反応速度の加速が妨げられることも説明できる。 以上本研究は、非対称なπ面をもつジベンゾビシクロ[2.2.2]オクタトリエン誘導体をデザインし、そのπ面選択特性を明らかにした。これらの基質のπ面は立体的には等価と考えられ、π軌道の非対称化と試薬の軌道の種類が反応面を決定していると考えられる。 | |
| 審査要旨 | ケトンや二重結合のπ結合が関与する反応においては、試薬の攻撃の方向は、多くの場合周辺の立体的な環境の大小によって決定される。しかし、この古典的立体化学によらない場合でもπ結合の反応面に非対称的な攻撃がおこる場合がある。岡本はこの立体効果によらないπ面の非対称化を実験的に示し、それを決定する理論を展開した。 岡本はジベンゾビシクロ[2.2.2]オクタトリエン骨格をもつ無水マレイン酸に対するDiels-Alder反応の面選択性を精査した。この骨格のベンゼン環上の置換基はこの反応点に対して単純な立体的な効果は及ぼさない。しかるに置換基によってはπ面の選択性が3:1にも及ぶことを示した。  このような選択性はフラノン誘導体に対するチオールの求核的1,4-共役付加においても観察されることを示した。この選択性も置換基の単なる立体的な大きさとか、電荷によっては説明できない。  岡本はこれらのπ面選択性を分子軌道法によって説明した。すなわち、ベンゾ環と置換ベンゾ環のπ*結合と反応中心π*結合の同位相の相互作用の大きさによって、第一次的にπ面選択性が決定する。Diels-Alder反応の場合には、ジエンのπ軌道の裏側(逆符合)がベンゾ部分のπ*と反結合的な相互作用が働き第一次の選択性を逆転させる。 この考え方はケトンのヒドリド還元やオレフィンの求電子的酸化反応にも適用できることが明らかになりつつあり、岡本の研究はπ面選択性の理論の拡大と一般化に寄与するものである。この成果は有機化学における立体化学研究にあたらしい展開をもたらすものであり、博士(薬学)に値するものである。 | |
| UTokyo Repositoryリンク | http://hdl.handle.net/2261/54594 |