本研究は、生体内での各種炎症の制御において重要な役割を演じていると考えられる肥満細胞の細胞外マトリックスへの接着の制御機構を明らかにするため、マウス骨髄由来肥満細胞(BMMC)を用いて高親和性IgE受容体(Fc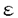 RI)を抗原特異IgEとその特異抗原で架橋することにより、細胞外マトリックスであるフィブロネクチン(FN)への接着を誘導し、その接着に関わるシグナルの解析を試みたものであり、下記の結果を得ている。 RI)を抗原特異IgEとその特異抗原で架橋することにより、細胞外マトリックスであるフィブロネクチン(FN)への接着を誘導し、その接着に関わるシグナルの解析を試みたものであり、下記の結果を得ている。 1. BMMC上のFcRIを抗DNP-IgE抗体とDNP-BSAにより架橋すると、現在までに報告されているホルボールエステル(PMA)やSteel factor(SLF)による刺激と同程度に細胞外マトリックスであるフィブロネクチン(FN)に一過性の接着を示した。この接着はインテグリンのVLA-5依存性であり、VLA-5の発現の上昇を伴わなかった。また、同じくFcRIの架橋によっておこるBMMCの脱顆粒の2次的効果ではなかったため、FcRIの架橋によりVLA-5の接着性自体が一過性に上昇したものと考えられた。 2. この接着は、SLF刺激による接着とほぼ同様に、チロシンキナーゼの阻害剤であるゲニステインにより抑制された。さらにPMA前処理によるプロテインキナーゼC(PKC)の抑制またはPI3-キナーゼの阻害剤であるウォルトマニン単独では十分な接着の阻害が見られなかったが、両者の併用によりほぼ完全に接着の阻害が見られたことより、この接着へのチロシンキナーゼやPKC、PI3-キナーゼの関与が示唆された。 ここで、FcRIの架橋によって活性化される非レセプター型チロシンキナーゼBtkについて、その変異体であるXIDマウス、Btk欠損マウスを用いてFcRIの架橋によるBMMCの接着への関与を調べた。その結果これらのマウス由来のBMMCではFcRIの架橋によるFN接着は野生型と差がなかったが、同じくFcRIの架橋による脱顆粒は変異マウス由来の肥満細胞で低下しており、FcRIの架橋による細胞内カルシウムの上昇についても変異マウス由来肥満細胞で低下が見られた。 3. 肥満細胞では、活性化に伴い細胞内カルシウムの上昇が起こることが知られている。接着をおこす刺激について細胞内カルシウムの変化を測定したところ、既に知られているFcRIの架橋に加えてSLF刺激によっても細胞内カルシウムの上昇が得られた。次に細胞内カルシウムの上昇を起こすイオノフォアを用いて接着の誘導を試みたところ、弱いながらもBMMCのFN接着が得られた。このことから、細胞内カルシウムの変動がBMMCの接着に1部関与している可能性が示唆された。 4. さらに、蛋白の脱リン酸化の接着への関与をprotein phosphatase1型と2A型(PP1,PP2A)の阻害剤であるオカダ酸とカリクリンAを用いて調べたところ、これらの阻害剤によりBMMCのFN接着は著明に阻害され、その濃度効果よりPP1の活性化の接着への関与が示された。PP2Bであるカルシニューリンについては、その阻害剤であるシクロスポリンAによりBMMCの脱顆粒は抑制されるにも関わらず、FN接着は有意に上昇し、その持続時間も延長した。この結果より、BMMCの各種刺激による接着性の抑制にカルシニューリンが関与している可能性が考えられた。 以上、本論文は骨髄由来肥満細胞上のインテグリンVLA-5のフィブロネクチンへの接着を誘導する新たな機構を報告し、その接着の制御機序について検索した。肥満細胞の液性因子の産生や増殖は細胞外マトリックスへの接着で増強することが既に報告されており、接着の制御機構を知ることは、これらの肥満細胞の機能の制御機序の解明に貢献をなすと考えられ、学位の授与に値するものと考えられる。 | 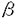 -glucosaminidaseの活性の比をp-nitrophenyl-N-acetyl
-glucosaminidaseの活性の比をp-nitrophenyl-N-acetyl