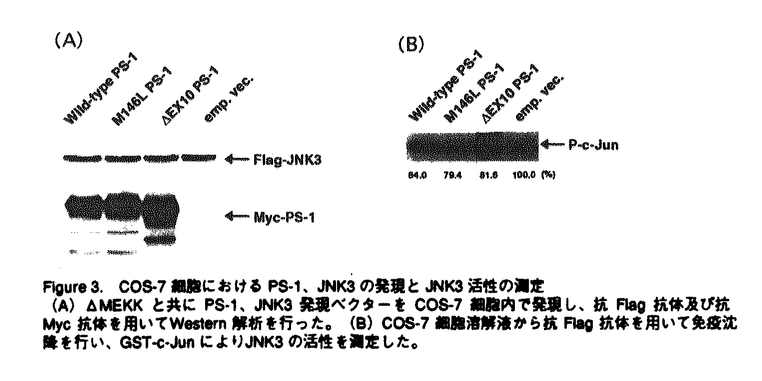
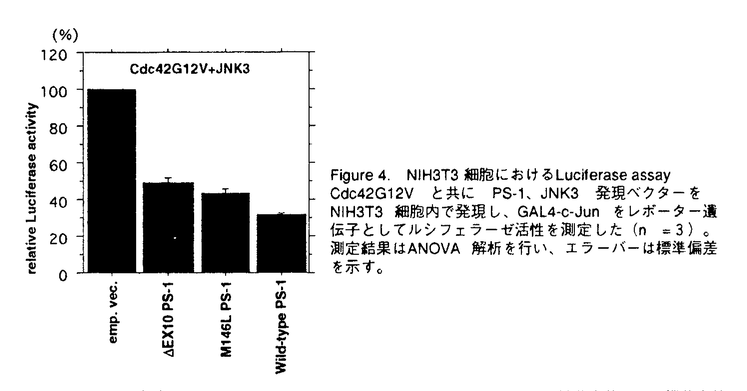
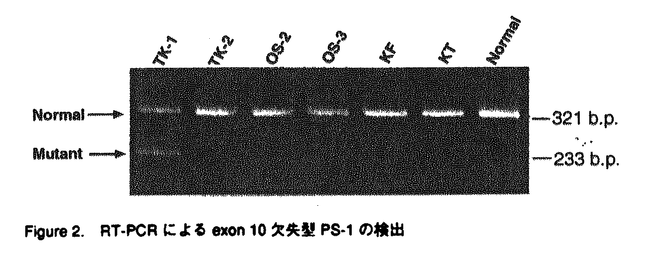
アルツハイマー病(AD)は、進行性の記憶障害や痴呆を主症状とする、高齢者の痴呆の原因として最も頻度の高い神経変性疾患である。AD患者脳に特徴的な病理学的変化としては、大脳皮質や海馬での神経細胞の脱落、 -アミロイド(A)を主成分とする老人斑、神経原繊維変化の出現などが知られるが、一方ではAD発症の分子メカニズムは不明とされている。本論文では、一般的なADの発症機構を明らかにする目的で、単一遺伝子疾患とされる早期発症型家族性AD(EOFAD)をモデルに、原因遺伝子の探索と遺伝学的解析、および分子生物学的手法を用いた機能解析を行っている。 -アミロイド(A)を主成分とする老人斑、神経原繊維変化の出現などが知られるが、一方ではAD発症の分子メカニズムは不明とされている。本論文では、一般的なADの発症機構を明らかにする目的で、単一遺伝子疾患とされる早期発症型家族性AD(EOFAD)をモデルに、原因遺伝子の探索と遺伝学的解析、および分子生物学的手法を用いた機能解析を行っている。 本論文は全3章から構成されている。第1章ではAD研究のこれまでの知見について概説した後、EOFADの原因遺伝子座とされる第14染色体q24.3領域にYAC contig mapを作成し、原因候補遺伝子の単離後に、本邦に存在するEOFAD患者を対象にした変異スクリーニングを行っている。一連の解析中に、Presenilin-1(PS-1)と名付けられた原因遺伝子がSherringtonらによって報告されたため、本申請者もこの報告に基づき本邦のEOFAD7家系について、同遺伝子の解析を行った。その結果、4家系から3種の新規点変異(Val96Phe、His163Arg、Ile213Thr)と1種の既知点変異(Ala260Val)、さらに1家系からはintron9のacceptor siteの塩基置換に起因する新規のabnormal splicing( EX10)変異を検出した。さらに、Val96Phe、His163Arg、Ile213Thr、EX10変異については、各家系の同胞について調査を行い、PS-1変異が各家系での発症原因であることを明らかにした。 EX10)変異を検出した。さらに、Val96Phe、His163Arg、Ile213Thr、EX10変異については、各家系の同胞について調査を行い、PS-1変異が各家系での発症原因であることを明らかにした。 -アミロイド蓄積の分子メカニズムは依然不明であったことや、第1章においてPS-1がEOFADの原因遺伝子であることが明らかになった事を受け、第2章ではPS-1が持つ生理機能の理解を分子生物学的手法により試みている。実験材料には、PS-1遺伝子を安定に保持し、且つ誘導可能な神経芽細胞腫株を樹立することで、これを培養細胞系でのモデルとしている。また、FAD変異型PS-1を保持する細胞株では、Aの産生が増加している事を見い出した。これらの細胞株を用いて、蛍光ディファレンシャルディスプレイ(FDD)法により、PS-1の発現によって特異的に挙動変化を示す遺伝子を探索した。合計411primer combinationを用いて、FAD変異型PS-1の誘導によって影響を受ける遺伝子を検索したところ、4種の遺伝子[immunoglobulin-binding protein(Bip)、induced PS-1 gene、2種のミトコンドリア遺伝子]を報告している。Bipは小胞体においてシャペロン蛋白として機能し、-アミロイド蛋白前駆体(APP)と結合してAPP蛋白の成熟やAの産生を抑制するという報告に注目した上で、PS-1の誘導によりBip mRNAが挙動変化を示したことは、APPの代謝機構の点からも興味深いが、現時点では両者の関係についての詳細は不明であると論じている。 第2章におけるFDD解析の結果から、FAD変異型PS-1が細胞内の全遺伝子の発現パターンに与える影響はわずかである事が判明した。またPS-1変異がA産生の増加をもたらすことは、ADの病理学的特徴を説明する上で理に叶うものであったが、PS-1の生理的機能の解明という点では他方面からの解析の必要性を論じている。そこで第3章では、PS-1のホモログであるPS-2がFas-ligandで誘導されたアポトーシスに対して抑制的に働くという報告をもとに、Fas-ligandで活性化が引き起こされるJNK pathwayとPS-1の関係に注目している。さらに、JNKファミリーの1つであるJNK3は神経特異的な発現パターンを示し、脳海馬においてアポトーシスを仲介することから、とくにJNK3を中心に報告している。培養細胞系において、カスケード上の構成的活性型であるCdc42G12VやMEKKを用いてJNK pathwayを活性化し、野性型およびFAD変異型PS-1によるJNK3活性化への影響を検討した。その結果、野性型PS-1はJNK3の活性化に抑制的に働くのに対し、FAD変異型PS-1ではその抑制能が減少していることを明らかにした。この結果から、アポトーシスを誘導するJNK pathwayの活性化機構にはPS-1が関与するという全く新しい概念を提唱し、AD発症の分子機構の解明に大きな貢献をした。 なお、本論文第1章は紙野晃人氏、三木哲郎氏、土井章良氏、伊井邦雄氏、P.H.St George-Hyslop氏、荻原俊男氏、榊佳之氏との共同研究であるが、論文提出者が主体となって分析及び検証を行ったもので、論文提出者の寄与が十分であると判断する。 したがって、博士(理学)の学位を授与できると認める。 | 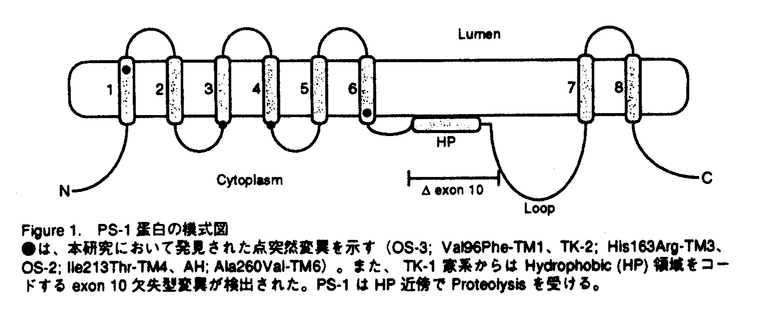
 -32PATPをインキュベーションし、リン酸化GST-c-Junの放射活性を測定・定量化した。その結果、野性型PS-1は
-32PATPをインキュベーションし、リン酸化GST-c-Junの放射活性を測定・定量化した。その結果、野性型PS-1は