材料設計の目的は、データベースやシミュレーショオンを用いて、要求される特性を満足する新材料を作り出すための構成元素、構造を見つけ出すことにある。材料の構造、化学元素とこれによって形成される物質系の特性間の関係を発見し、特定の性質を有する材料を生産するためのプロセス手法を設計することも、材料設計に課せられた大きな課題の一つである。物性を支配する様々な支配要因の内、微視的な原子構造と組成は、最も基本で重要なものである。酸化物の持つ物性の多様性は以前から注目され、最近ではこれを利用した新デバイス開発に関する研究が盛んに行われている。シミュレーションもが統合された物質データベースを利用した材料設計の応用が特にこの分野にも期待されていることは言うまでもない。透明導電性酸化物はその高い電気伝導性と可視光透過性から、多くの光電子デバイスに使用されている。 太陽電池、フラットパネルディスプレイ用の透明電極、熱性反射硝子用のヒートミラーなどがその代表例である。In2O3にSnをドープしたITOは、透明導電物質の代表であり現在でも工業的に盛んに使用されている。しかしながら、Inのコストと残存資源量の問題からITOに変わる新しい透明導電性物質の開発が望まれている。ZnOにAl、Ga、Inをドープした物質は、このITOの代替物質として注目され、特に太陽電池用の透明電極として大きく期待されている。 このような産業的な要請から、これまでに様々な手法により、この物質の構造と物性間の関係定量化に関する研究が活発に進められてきている。本研究では、最近発展の目覚しい第一原理計算を用いて、このZnOを母体とした透明導電性物質の特徴を見出し、格子欠陥構造と電子構造関係を解明をすることを目標とした。まず第一に、ZnOと他の透明導電性物質の母材結晶SnO2,In2O3のバンド構造を比較することによって、ZnOの特徴を評価した。電子構造意外にも、バンド間遷移による複素誘電率を計算することから、紫外線吸収端のエネルギー依存性を評価し、他物質との違いにとその工業的利用価値について検討した。次に、ZnO中の格子欠陥構造とその電子構造、エネルギー的安定性について研究した。また最後に、キャリアとイオン化不純物間へのモデルポテンシャルの導入に基づき、新たなイオン化不純物散乱によって起こした支配されるキャリアの移動度および電気伝導率の定式化について解析式を提案し、その具体的な形を導いた。 電子構造解析には、密度汎関数法に基づく第一原理計算を用いた。内核電子+原子核の価電子への寄与を記述するために、KleinmannとBylanderによって提案された分離型ノルム保存型の擬ポテンシャル法を使用した。電子交換相互作用については、Ceperley-Alder型の局所密度近似を用いた。一電子波動関数は、基底関数として平面波を用い展開し、前処理付き共役勾配法によって全エネルギーを直接的に最小化することで、密度汎関数法に基づく電子系の基底状態を求めた。原子位置の最適化に関しては、Broyden-reduced Goldfarb-Shannon atomicrelaxation methodを用いた。バンド間遷移による複素誘電率の計算は、その虚数部を  実数部に関しては、虚数部 2をKramers-Kronig変換することによって求めた。 2をKramers-Kronig変換することによって求めた。 図1にZnO,SnO2,In2O3のバンド構造を示す。ZnOの価電子帯は他物質に比較して、二つに分裂していることが特徴的である。これは主にZnの3dレベルが、In,Snの4d,レベルより高い位置に存在することから説明できる。伝導帯はどの物質においても価電子帯よりも高い分散を示す。この結果は、実験的に観測されるキャリアドープの際の光学的バンドギャップの増大機構、つまりBurstein-Moss shift(7)を説明している。また、この結果はこれらの物質がn型伝導を主とする導電物質になりやすいことを示している。(格子欠陥の自己補償効果などが原因による単極性が原因で、これら物質の電気伝導性が支配されていることも考えられる。)計算によって得られたバンドギャプは、ZnO,SnO2,In2O3に関してそれぞれ0.8,0.7,0.9eVと実験値である3.4,3.6,3.7eVと比べてかなり小さいがこれは局所密度近似に基づくものである。本研究では、複素誘電率の波長依存性を評価する上で、この問題を伝導帯をバンドギャップの過小評価分だけ上方にシフトさせている。図2に各偏光光線に対する複素誘電率を示す。直接この物理量に関する実験値が存在しなかったため、複素誘電率を光学定数に換算し、その実験結果と比較したものを図3に示す。この結果、本研究で採用された計算法は高い精度で実験値を再現していることが分かり、実験的なデータの存在しない複素誘電率の偏光成分についても同様な評価精度が得られていることが期待できる。ZnOはその結晶構造の異方性から、[001]つまりC軸方向に偏向した光に対して大きな透過性を示し、SnO2でもこの事情は同様である。スパッタ方による真空成膜において、ZnOはガラス基板上にC軸ハ配向することがあり、このようなプロセスと結び付けることによって、一定の偏向光線によって高い透過性ももつ透明導電膜が作成できる可能性がある。このようにIn2O3と比較し、光学異方性を示す物質として、ZnOの工学的用途が特徴付けられた。この外、本論文中では、ZnO,SnO2、In2O3の化学結合状態の違いについても検討されている。本研究では、透明導電物質の光学、電子的特性が第一原理的手法に基づき研究された。特にITO代替物質として注目されているZnOに関する電子構造、光学特性がIn2O3,SnO2と比較されながら評価された。また局所密度近似に基づく光学特性の評価法の有効性についても確認された。 キャリアとイオン化不純物間へのモデルポテンシャルの導入に基づき、Boltzmann輸送方程式における緩和時間近似および確率的にイオン化不純物のドーピングされた材料中に行われるキャリアとのイオン化不純物の弾性散乱を考えて、退化または非退化の場合でのイオン化不純物の散乱によって起こしたキャリアの移動度について解析式を提案し、その具体的な形を導いた。その解析式は以下のようである。 非退化の場合 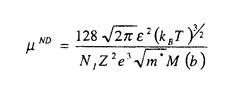 ただし、  N1はイオン化不純物の密度ただし、nはキャリアの密度ただし、 はキャリアの有効質量ただし、は誘電率である。 はキャリアの有効質量ただし、は誘電率である。 退化の場合 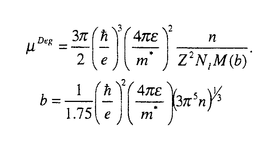 ここで、 これらの式は物理的な合理性を持ち、実用的だと考えられる。現在よく使わるBrooks-Herringの式を用いた場合での移動性の過大評価に対して改善することができる。これらの式を用いた計算結果は実験結果とよく一致したことによって(テーブル1)、キャリアの移動度に関するこれらの式の記述はBrooks-Herringのものと比べて優れていると言える。 第一性原理に基づく総エネルギー擬ポテンシャル法とスーパーセル計算法の併用及びキャリアの移動度についての評価により、アルミニウムのドーピングされた酸化亜鉛(ZnO: l)が持っている高い電気伝導率は格子欠陥のイオン化状態によるものであることが分かった。従って、ZnO:lの高い電気伝導率を導いた多数の格子欠陥中の支配的な因子は l)が持っている高い電気伝導率は格子欠陥のイオン化状態によるものであることが分かった。従って、ZnO:lの高い電気伝導率を導いた多数の格子欠陥中の支配的な因子は の格子欠陥とそのイオン化状態だと推測できる。こういったことから、アルミニウムのドーピングされた酸化亜鉛の高い電気伝導率の存在に関する物理的なメカニズムを解明した。 の格子欠陥とそのイオン化状態だと推測できる。こういったことから、アルミニウムのドーピングされた酸化亜鉛の高い電気伝導率の存在に関する物理的なメカニズムを解明した。 今までの研究結果は、密度の汎関数において第一性原理に基づく総エネルギー擬ポテンシャル法とスーパーセル法の併用がドーピングされたシステムの解析に対して大変有効であることが実証した。  (図1)(A)ZnO(B)SnO2(C)In2O3 (図1)(A)ZnO(B)SnO2(C)In2O3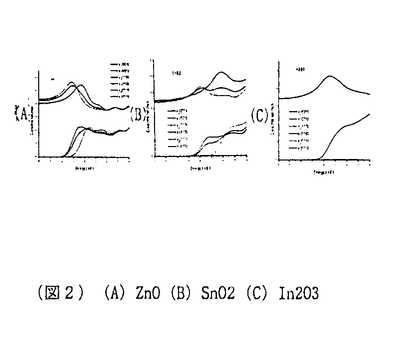 (図2)(A)ZnO(B)SnO2(C)In2O3 (図2)(A)ZnO(B)SnO2(C)In2O3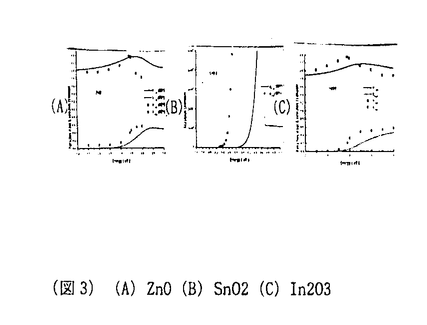 (図3)(A)ZnO(B)SnO2(C)In2O3 (図3)(A)ZnO(B)SnO2(C)In2O3 テーブル1 テーブル1 |