ノシセプチン(オーファニンFQ)はオピオイド受容体様オーファンレセプターの内在性のリガンドであると最近同定された17個のアミノ酸残基からなる神経ペプチドである。しかし、その機能についてはほとんどわかっていない。そのため、ノシセプチン受容体(オピオイド受容体様オーファンレセプター)のN末端側約1/3をコードするエクソンを欠損させ、代わりに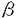 -ガラクトシターゼとネオマイシン耐性遺伝子を5’非翻訳領域に挿入した人工変異を導入することでノシセプチン受容体を欠損したマウスが作製され機能解析が行われた。まず、Morrisの水迷路学習および受動的回避学習の行動実験においてノシセプチン欠損マウスは、野生型マウスより学習、記憶に関する能力が向上していることが明らかになった。次に、-ガラクトシターゼ染色法およびin situ hybridization法により、ノシセプチン受容体およびノシセプチン前駆体が、学習、記憶において重要な役割を果たすことが知られている海馬に発現していることが示された。海馬の興奮性シナプスにおいて入力に高頻度刺激を与えると、シナプス伝達の長期増強(LTP)が誘導されることが知られており、LTPは学習や記憶の細胞レベルでのモデルとされている。本研究では、ノシセプチンの中枢神経系における役割を解明するため、変異型、野生型双方より作製した海馬スライス標本を用いてシナプス伝達および可塑性におけるノシセプチン受容体の機能を検討した。 -ガラクトシターゼとネオマイシン耐性遺伝子を5’非翻訳領域に挿入した人工変異を導入することでノシセプチン受容体を欠損したマウスが作製され機能解析が行われた。まず、Morrisの水迷路学習および受動的回避学習の行動実験においてノシセプチン欠損マウスは、野生型マウスより学習、記憶に関する能力が向上していることが明らかになった。次に、-ガラクトシターゼ染色法およびin situ hybridization法により、ノシセプチン受容体およびノシセプチン前駆体が、学習、記憶において重要な役割を果たすことが知られている海馬に発現していることが示された。海馬の興奮性シナプスにおいて入力に高頻度刺激を与えると、シナプス伝達の長期増強(LTP)が誘導されることが知られており、LTPは学習や記憶の細胞レベルでのモデルとされている。本研究では、ノシセプチンの中枢神経系における役割を解明するため、変異型、野生型双方より作製した海馬スライス標本を用いてシナプス伝達および可塑性におけるノシセプチン受容体の機能を検討した。 成熟マウス(5〜9週齢)より作製した厚さ400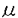 mの海馬スライス標本においてシャッファー側枝を0.1Hzで電気刺激して誘発した興奮性シナプス後電位(EPSP)を細胞外電位記録法を用いてCA1領域の放線状層から記録した。まず、ノシセプチン受容体欠損のシナプス伝達効率への影響を検討した。細胞外電位記録法によりEPSPを測定する際、その直前に小さな下向きの成分(fiber volley)が見られるが、これは入力線維の軸索に生じた活動電位を反映しており、その大きさは刺激されている線維の数に比例する。よって、このfiber volleyの大きさ及びEPSPの大きさをそれぞれシナプス伝達の入力及び出力の指標とすることで、両者間の関係を野生型と変異型で比較した。通常の条件下ではEPSPの大きさに対してfiber volleyの大きさは相対的に極めて小さいため、正確にその比を求めることは難しい。そこで、海馬CA1領域の興奮性シナプスにおけるシナプス伝達を媒介するAMPA受容体のアンタゴニスト、CNQXを低濃度にて実験中灌流させることで、シナプス後細胞における感受性を低下させ、EPSPの大きさがfiber volleyの大きさと同程度になるようにしてそれぞれの値を測定した。この条件下で刺激強度を上昇させていき、fiber volleyの大きさに対してEPSPの大きさをプロットして、その回帰直線の傾きを比較したが、野生型、変異型間でこの傾きの値に有意な差は認められなかった。このことから、ノシセプチンは正常状態でのシナプス伝達効率の調節には関与していないことが示唆された。 mの海馬スライス標本においてシャッファー側枝を0.1Hzで電気刺激して誘発した興奮性シナプス後電位(EPSP)を細胞外電位記録法を用いてCA1領域の放線状層から記録した。まず、ノシセプチン受容体欠損のシナプス伝達効率への影響を検討した。細胞外電位記録法によりEPSPを測定する際、その直前に小さな下向きの成分(fiber volley)が見られるが、これは入力線維の軸索に生じた活動電位を反映しており、その大きさは刺激されている線維の数に比例する。よって、このfiber volleyの大きさ及びEPSPの大きさをそれぞれシナプス伝達の入力及び出力の指標とすることで、両者間の関係を野生型と変異型で比較した。通常の条件下ではEPSPの大きさに対してfiber volleyの大きさは相対的に極めて小さいため、正確にその比を求めることは難しい。そこで、海馬CA1領域の興奮性シナプスにおけるシナプス伝達を媒介するAMPA受容体のアンタゴニスト、CNQXを低濃度にて実験中灌流させることで、シナプス後細胞における感受性を低下させ、EPSPの大きさがfiber volleyの大きさと同程度になるようにしてそれぞれの値を測定した。この条件下で刺激強度を上昇させていき、fiber volleyの大きさに対してEPSPの大きさをプロットして、その回帰直線の傾きを比較したが、野生型、変異型間でこの傾きの値に有意な差は認められなかった。このことから、ノシセプチンは正常状態でのシナプス伝達効率の調節には関与していないことが示唆された。 正常クレブス液灌流下で安定なEPSPを記録した後、高頻度刺激(100Hz,1秒)を与えると、変異型、野生型双方において60分以上にわたるLTPが誘導された。しかし、変異型マウスでは野生型マウスに比べ約2倍の大きさのLTPを示した。この結果からノシセプチンは海馬CA1領域でのLTP形成に抑制的に作用していることが示唆された。ノシセプチンは海馬の神経伝達物質放出に関与しているNおよびP/Q型カルシウムチャネルを抑制すると報告されていることから、変異型マウスでは、高頻度刺激の際により多くのグルタミン酸が放出され、これによりLTP誘導に必要なグルタミン酸受容体がより強く活性化されLTPが促進した可能性がある。しかしシナプス前末端の伝達物質放出確率に関連する短期可塑性、1)短い間隔で2回連続して刺激を行うことで誘導されるEPSPの促進(paired-pulse facilitation:PPF)2)NMDA受容体アンタゴニスト存在下で高頻度刺激により誘導される増強(post-tetanic potentiation:PTP)は、変異型と野生型の間で差異は認められなかった。以上から、変異型マウスにおいては、伝達物質放出機構に異常はなく、高頻度刺激の際により多くのグルタミン酸が放出されたためにLTPが促進した可能性は低いと思われる。また、ノシセプチンは海馬のCA1領域の錐体細胞においてG蛋白共役型内向き整流性カリウムチャネルとカップリングしているという報告があることから、変異型マウスでは、高頻度刺激の際の脱分極がより大きく、そのためNMDA受容体のより強い活性化が起こりLTPが促進される可能性もあるが、高頻度刺激中のシナプス後細胞脱分極の程度に差異が認められなかったため、脱分極の違いによりLTPの大きさに差が出たとは考えにくい。しかし、変異型マウスの錐体細胞の静止膜電位が野生型マウスよりも脱分極側に変化しているとすると、EPSPの駆動力が小さくなるため高頻度刺激中のシナプス後細胞脱分極を過小評価する可能性がある。これを検討するため、錐体細胞からホールセル記録にてグルコン酸カリウムを主成分とした細胞内液を用いて静止膜電位を測定したが、変異型と野生型の間に差異は認められず、この可能性は否定できる。以上から、野生型、変異型マウスにおけるシナプス前及びシナプス後細胞の電気生理学的特性の違いからLTPの大きさに差が出たとは考えにくい。 ノシセプチンが内在性ペプチドとして同定されて以来、ノシセプチン投与により様々な応答が誘導されることが報告されているが、現在までノシセプチン受容体(オーファンレセプター)のアンタゴニストが開発されていないため、本当にこれらの応答がノシセプチン受容体を介する反応であるかは不明であった。ノシセプチンが海馬のCA1領域の錐体細胞の内向き整流性カリウムチャネルを活性化させるという性質を利用して、錐体細胞からホールセル記録を行い膜電位を-70mVに電位固定した状態でノシセプチンにより誘導される膜電流を観察した。野生型マウスにおいては外向き電流が認められたが、変異型マウスにおいては認められなかった。このことから、本研究で用いたノシセプチンはノシセプチン受容体のリガンドとして作用しており、ノシセプチンはノシセプチン受容体(オーファンレセプター)のアゴニストであることが生理学的に初めて示された。 次にノシセプチンを3分間投与した直後に高頻度刺激を行ったところ、誘導されたLTPの大きさはノシセプチン非存在下で誘導されたLTPの大きさと同程度のものであった。このことは、高頻度刺激の際に放出されるノシセプチンの量は充分であるためLTP発現の修飾に必要な量に到達しており、そのため外部からのノシセプチンによる影響は受けないということで説明できるかもしれない。あるいは、高頻度刺激においてはシナプスより放出されたノシセプチンの影響は受けない抑制性介在ニューロンがノシセプチン灌流投与により影響を受け、錐体細胞への抑制的な要素が抑えられ、ノシセプチンの錐体細胞への直接的な抑制作用と拮抗するという可能性も考えられる。 本研究においてノシセプチン受容体が海馬CAI領域でのLTP誘導において抑制的な役割を担っていることが示唆された。しかし、ノシセプチン受容体欠損マウスにおけるLTP促進機構についてはいまだ解明されていない。本研究の結果からはそのメカニズムとして、変異型マウスでのシナプス後細胞におけるシグナル伝達系の変化が候補として挙げられる。ノシセプチン受容体が活性化されることにより環状AMP生成が抑制されることから、高頻度刺激により放出されたノシセプチンはシナプス後細胞における環状AMPのレベルの調節を介してLTP形成に抑制的に作用している可能性がある。したがって、変異型マウスでみられるLTPの促進は、ノシセプチン受容体を欠損させたことによる環状AMP生成の抑制的調節の欠如が原因かもしれない。また、プロテインキナーゼCおよびMAPキナーゼといったキナーゼの活性化も、ノシセプチン受容体が活性化されることにより誘導されることから、ノシセプチン受容体を欠損させた結果、プロテインキナーゼCおよびMAPキナーゼ系の異常が変異型マウスでのLTPの亢進を誘発する可能性も考えられる。 |