アポB欠損症は、先天的にアポBおよびアポB含有リポ蛋白が欠損する病態であり、著明な低脂血症をきたす。アポB欠損症の病因を分子遺伝学的に明らかにすることは、その的確な診断と治療に不可欠であるのみならず、アポB含有リポ蛋白の合成分泌過程の解明、ひいては高脂血症の新たな治療法の開発に通じる重要な意義を持つ。本研究は、アポB欠損症が疑われた3症例の臨床像を詳細に記述し、その分子遺伝学的解析を行ったものであり、下記の結果を得ている。 1.アポB欠損症には、遺伝型式の異なる無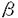 リポ蛋白血症(abetalipoproteinemia;ABL)と家族性低リポ蛋白血症(familial hypobetalipoproteinemia;FHBL)がある。FHBLは常染色体共優性遺伝性疾患であり、1987年、異常な短縮型アポBの発見を契機にアポB遺伝子の異常が同定され、アポB遺伝子の異常によって起こることが明らかになった。一方、ABLは常染色体劣性遺伝で、その原因遺伝子は長らく不明であった。しかし、1993年になって本症でミクロソームトリグリセライド転送蛋白(MTP)遺伝子の変異が同定され、アポB含有リポ蛋白の合成過程におけるMTPの重要性が注目されている。両疾患ともホモ接合体では著明な低脂血症をきたすが、ABLのヘテロ接合体は脂質レベル正常、FHBLでは中程度の低脂血症を示す。申請者はまず、遺伝子レベルで両疾患を鑑別するために、MTPおよびアポB各遺伝子について各々の多型性マーカーを用いてハプロタイプ解析を試みている。すなわち、MTP遺伝子イントロン10のCA repeatおよびアポB遺伝子の3’VNTRを含む領域をそれぞれの多型性マーカーとしてPCRで増幅し検討した。2例はMTPがホモ、アポBはヘテロであり、臨床像と合わせMTP遺伝子異常の存在が示唆された。逆にもう1例はMTPがヘテロ、アポBがホモであり、アポB遺伝子異常が疑われた。 リポ蛋白血症(abetalipoproteinemia;ABL)と家族性低リポ蛋白血症(familial hypobetalipoproteinemia;FHBL)がある。FHBLは常染色体共優性遺伝性疾患であり、1987年、異常な短縮型アポBの発見を契機にアポB遺伝子の異常が同定され、アポB遺伝子の異常によって起こることが明らかになった。一方、ABLは常染色体劣性遺伝で、その原因遺伝子は長らく不明であった。しかし、1993年になって本症でミクロソームトリグリセライド転送蛋白(MTP)遺伝子の変異が同定され、アポB含有リポ蛋白の合成過程におけるMTPの重要性が注目されている。両疾患ともホモ接合体では著明な低脂血症をきたすが、ABLのヘテロ接合体は脂質レベル正常、FHBLでは中程度の低脂血症を示す。申請者はまず、遺伝子レベルで両疾患を鑑別するために、MTPおよびアポB各遺伝子について各々の多型性マーカーを用いてハプロタイプ解析を試みている。すなわち、MTP遺伝子イントロン10のCA repeatおよびアポB遺伝子の3’VNTRを含む領域をそれぞれの多型性マーカーとしてPCRで増幅し検討した。2例はMTPがホモ、アポBはヘテロであり、臨床像と合わせMTP遺伝子異常の存在が示唆された。逆にもう1例はMTPがヘテロ、アポBがホモであり、アポB遺伝子異常が疑われた。 2.MTP遺伝子異常が疑われた2例につき、MTP遺伝子の全エクソンおよびエクソン-イントロン接合部についてPCR法により全塩基配列を決定し、それぞれ異なる変異を同定した。1例は、エクソン11、cDNA1385-1389の5連続のアデニンの一つが欠失するフレームシフト変異で、活性に重要なC末端領域を完全に欠如した不完全な蛋白が生成されるものと考えられた。もう1例は、エクソン16、cDNA2338番目のA→T変異によりAsn780がTyrに置換されるミスセンス変異であった。我が国のABL症例としては遺伝子変異が同定されたのははじめてのことであり、人種を越えてMTP欠損がABLの原因であることを明らかにした。 3.このミスセンス変異が実際にMTPを不活化させることを確認するために、COS-1細胞を用いた一過性発現系で検討している。正常あるいは本変異を導入したMTP発現ベクターを構築し、COS-1細胞にトランスフェクトして、細胞抽出液のMTP発現量と活性を比較した。イムノブロット上、変異MTPは正常MTPと同レベルの発現を認めるにも関わらず、活性はほとんど消失しており、本変異がMTP不活化の原因であることが証明された。未だ不明な点の多いMTPの機能ドメインを検討する上で重要な知見である。MTP遺伝子のミスセンス変異は世界で2例目であり、極めて貴重な報告となっている。 4.アポB遺伝子異常が疑われた症例では、アポ蛋白のSDS-PAGEにより約195kDaの異常な短縮型アポBを見い出した。これよりGln1755→stopとなるナンセンス変異を同定している。本変異の短縮型アポBはB38.7と命名された。本例はそのホモ接合体であったが、FHBLのホモ接合体は世界でこれまで数例の報告しかなく、やはり貴重な症例であった。 以上、本論文はアポB欠損症3例について臨床的な検討を行い、遺伝子レベルでその病因を同定している。2例はいずれも新たなMTP遺伝子変異による無リポ蛋白血症、1例はやはり新たなアポB遺伝子変異による家族性低リポ蛋白血症であった。これらの変異の同定は、アポB含有リポ蛋白の合成過程の解明に重要な貢献をなすものであり、それらの臨床像を詳細に記載した意義も大きい。 よって、学位の授与に値するものと認められる。 | 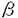 リポ蛋白血症と家族性低
リポ蛋白血症と家族性低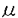 g/ml(正常下限)であった。内視鏡上、十二指腸粘膜は正常で、光顕レベルでは粘膜上皮細胞への脂質の蓄積は明かでなかったが、電顕では脂肪滴の存在が確認された。肝腫瘍は血管腫であった。また、腹部CT上、主要な動脈の著明な石灰化がみられた。本例は、家族に低脂血症を認めることから、家族性低
g/ml(正常下限)であった。内視鏡上、十二指腸粘膜は正常で、光顕レベルでは粘膜上皮細胞への脂質の蓄積は明かでなかったが、電顕では脂肪滴の存在が確認された。肝腫瘍は血管腫であった。また、腹部CT上、主要な動脈の著明な石灰化がみられた。本例は、家族に低脂血症を認めることから、家族性低