学位論文要旨
| No | 114562 | |
| 著者(漢字) | 出口,順夫 | |
| 著者(英字) | ||
| 著者(カナ) | デグチ,ジュンオ | |
| 標題(和) | 血管障害後の内膜肥厚におけるPDGF-B鎖の役割とその発現調節の解析 | |
| 標題(洋) | ||
| 報告番号 | 114562 | |
| 報告番号 | 甲14562 | |
| 学位授与日 | 1999.03.29 | |
| 学位種別 | 課程博士 | |
| 学位種類 | 博士(医学) | |
| 学位記番号 | 博医第1482号 | |
| 研究科 | 医学系研究科 | |
| 専攻 | 外科学専攻 | |
| 論文審査委員 | ||
| 内容要旨 | バルーン血管拡張術や動脈血行再建術後に発生する内膜肥厚は、再狭窄の一因となり、その病態解明および治療法の確立は急務である。内膜肥厚は、血管壁に加えられた障害により活性化された中膜平滑筋細胞が内膜に遊走し、増殖を繰り返すことにより形成されると考えられている。これらの過程には様々な増殖因子が関与していると考えられているが、十分に解明されていない。 血小板由来増殖因子(platelet-derived growth factor、以下PDGFと略す)は、内膜肥厚の病態において最も注目を集めている増殖因子である。PDGFにはA鎖とB鎖があり、ホモダイマーもしくはヘテロダイマーを形成している。A鎖はPDGF- アンギオテンシンII(AngII)変換酵素阻害剤やAngII-I型受容体(AT1R)拮抗薬の投与は、バルーン障害後の内膜肥厚の形成を強く抑制する。この結果は、AngIIが内膜肥厚の形成に関与していることを示している。しかし、AngIIがどのような機序により内膜肥厚の形成に関与しているのかは明らかになっていなかった。私たちは、最近、ラット頚動脈バルーン障害モデルにおいて、障害血管のPDGF受容体のチロシンリン酸化レベルがAT1R拮抗薬の投与により抑制されることをみいだしている。このことは、AngIIが障害血管局所においてPDGF-B鎖の発現調節に関わっている可能性を示している。 私は、本研究において第一に、アデノウイルスによる血管壁への遺伝子導入法を用いて、内膜肥厚におけるPDGF-B鎖の役割を直接的に検討した。この実験ではPDGF- PDGFXRをコードするcDNAを組み込んだ、複製能力の欠如したアデノウイルス(AdenoXR)を作成した。コントロールアデノウイルスとして大腸菌 ウイスターラット(オス、体重300g)の左総頚動脈にバルーン内皮剥離障害術を施行した。5日後に上記のウイルスを頚動脈内腔に投与した。バルーン障害後8、10、14日後に頚動脈を摘出し、(1)Xgal染色、(2)BrdU染色、(3)内膜中膜面積比(I/M比)、(4)ウエスタンプロットによるPDGF- PDGFXR cDNAを含む発現プラスミドpCAGGS-XRをCHO-K1細胞にトランスフェクトし、CHO細胞PDGFXR安定発現株を樹立した。その培養上清を採取後濃縮し、wheat germ agglutinin(WGA)Sepharose6MBにて精製した。一部の実験には昆虫細胞(Sf9)で産生した組換えPDGFXR蛋白を用いた。 バルーン障害頚動脈の内膜肥厚部分、生後7日令のラット大動脈、生後18週令のラット大動脈よりexplant法にて培養平滑筋細胞(SMC)を樹立した(各々、neointimal SMC,newborn rat SMC、adult rat SMCと略す)。 総RNAを抽出し,ホルムアルデヒドゲルにより泳動分離後,ナイロン膜に転写し,[ AngII(10-8M)の添加36時間後に,培養上清を採取し,トリクロロ酢酸を用いて上清中のタンパクを変性回収した.SDS-PAGEにて泳動分離後ナイロン膜に転写し,PDGF-B鎖を認識する特異抗体でウエスタンプロットした. 細胞溶解物をそれぞれの特異抗体で免疫沈降し,[ 約1.0kbのPDGF-B鎖プロモーター領域(-956〜+45)を,ルシフェラーゼレポーターベクターpGL2basicに組み込み、レポーターベクターSis-Lucを作製した。Sis-Lucを細胞にトランスフェクションし,AngII刺激4時間後に細胞を溶解し、ルシフェラーゼ活性を測定した. (1)AdenoXRをSMCに感染させ、RT-PCRにてPDGFXRmRNAの発現を確認した。ウエスタンプロット解析にて、PDGFXRは培養上清に認められ、細胞外に分泌されていることを確認した。AdenoLacZを感染させたSMCの培養上清には,PDGFXRは全く検出されなかった。 (2)AdenoXR感染SMCのPDGFXRの分泌量は、感染後2日目にほぼ最大になり一週間後でも最大値の40%の分泌が認められた。 (3)組換えPDGFXRをSMCの培養上清に加えると,PDGF-BBによるPDGF- (1)障害直後に比べ障害5日目にウイルスを感染させることにより、遺伝子導入効率が飛躍的に高まった。この条件で障害血管に遺伝子を導入した。AdenoXR投与群では、アデノウイルス非投与群(内膜剥離障害(+))に比較して内膜面積が抑制され、I/M比が43%に低下した。一方、AdenoLacZ投与群では、内膜面積およびI/M比はアデノウイルス非投与群(内膜剥離障害(+))と同程度であった。 (2)AdenoXR投与群では、PDGF- (3)AdenoXR投与群ではAdenoLacZ投与群に比較して、内膜細胞のBrdU取り込みは著しく減少していた。中膜細胞へのBrdU取り込みもAdenoXR投与群で低下傾向がみられた。 (1)neointimal SMCおよびnewborn rat SMCでは、AngII刺激によりPDGF-B鎖mRNAの発現が誘導された。これとは対照的に、adult rat SMCではPDGF-B鎖mRNAはAngIIによって誘導されなかった。また、AngII刺激によりnewborn rat SMCの培養上清中のPDGF-Bタンパクは増加した。 (2)newborn rat SMCにおける、AngIIによるPDGF-B鎖mRNAの発現誘導は、AT1R拮抗薬CV11974で完全に抑制されたが、AT2R拮抗薬PD123319では抑制されなかった。 (3)AngII刺激により3つのMAPキナーゼ(ERK,JNK,p38)のいずれも活性化された。MEK阻害剤であるPD98059は、AngIIによるPDGF-B鎖mRNAの発現をほぼ完全に抑制した。一方p38阻害剤であるSB203580には抑制作用はなかった。 (4)Sis-Lucをトランスフェクトしたnewborn rat SMCをAngIIで刺激すると、ルシフェラーゼ活性が上昇した。また、Sis-Lucとともに各MAPキナーゼの優性抑制型発現ベクターを遺伝子導入した。AngIIによるルシフェラーゼ活性の増加は優性抑制型ERKおよび優性抑制型JNKの発現により抑制された。優性抑制型p38には抑制作用はなかった。 (5)アデノウイルスを用いて優性抑制型Ras変異体を発現させると、AngIIによるERKの活性化とPDGF-B鎖mRNAの発現誘導は抑制された。しかしJNKの活性化は抑制されなかった。 (5)サイクリックGMPおよびサイクリックAMPの誘導体により、AngIIによるERK及びJNKの活性化は抑制され、これとともにPDGF-B鎖mRNAの発現も抑制された。 障害後5日目にアデノウイルスを投与することで遺伝子導入効率は飛躍的に上昇した。LacZ遺伝子は、ほぼすべての内膜細胞に導入された。一方、中膜平滑筋細胞はごく少数が遺伝子導入された。内膜と中膜に存在する平滑筋細胞間で、アデノウイルスによる遺伝子導入効率に差がある理由は現在のところ不明である。あるクラスのインテグリンはアデノウイルスの細胞への侵入経路として関与している可能性が報告されていることから、内膜細胞と中膜細胞に発現しているインテグリンのクラスの違いが感染効率の差異の原因の一つである可能性がある。 障害後5日目にPDGFXR発現ウイルスを感染させることにより、その後の内膜肥厚形成が著明に抑制された。従来、血管障害時に血小板より放出されたPDGFが内膜形成の早期の段階(障害後4〜5日以内)で関与しているとされていたが、本実験の結果は、内膜が急速に増大する時期(5〜14日)においてもPDGF-B鎖が中心的な働きをしていることを示している。さらにこの結果は血小板の影響の少なくなった同時期において、PDGF-B鎖が血管壁自体で産生されていることを示唆している。また、PDGFXRが、in vivo血管においてPDGF-B鎖の作用を十分に阻害し得ることが示されたことから、PDGFXRはPDGF-B鎖が関与する他の疾患において有力な治療手段となり得ることが予想される。 従来より肥厚内膜を形成する平滑筋細胞は、成体型より幼若型の平滑筋細胞に形質が類似していることが指摘されていた。今回、neointimal SMCとnewborn rat SMCにおいてのみAngII刺激でPDGF-B鎖の発現がみられたことは、形質の転換した細胞が、局所でオートクリン、パラクリン様式の増殖機構を有していることを示唆している。 内因性PDGF-B鎖が内膜肥厚の形成において重要な働きをしていることが明らかとなった。さらに、PDGF-B鎖は内膜平滑筋細胞で産生され、アンギオテンシンIIによる調節を受けていることが示唆された。 | |
| 審査要旨 | 血小板由来増殖因子(platelet-derived growth factor、PDGF)は、動脈損傷後に発生する内膜肥厚において重要な役割を演じていると考えられていたが、最近、内膜肥厚が急速に進展する時期に一致してPDGF受容体が強く活性化されているということが明かとなった。本研究では、ラットバルーン障害モデルを用いて、内膜肥厚進展期におけるPDGFの役割を直接的に解析し、さらにその発現調節について検討を加えたものであり、下記の結果を得ている。 PDGF- (1)ラット頚動脈バルーン障害後の血管壁への遺伝子導入効率を、大腸菌 (2)AdenoXR投与群では、アデノウイルス非投与群(内膜剥離障害(+))に比較して内膜面積が抑制され、内膜面積中膜面積比(I/M比)が43%に低下した。一方、AdenoLacZ投与群では、内膜面積およびI/M比はアデノウイルス非投与群(内膜剥離障害(+))と同程度であった。 (3)AdenoXR投与群ではPDGF- (4)AdenoXR投与群ではAdenoLacZ投与群に比較して、内膜細胞のBrdU取り込みは著しく減少していた。中膜細胞へのBrdU取り込みもAdenoXR投与群で低下傾向がみられた。 内膜平滑筋細胞neointimal SMCは、成体ラット中膜平滑筋細胞Adult rat SMCsと形態的および遺伝子発現上形質が異なっており、むしろ幼若ラット中膜平滑筋細胞newborn rat SMCに近いことがわかってきた。従って、neointimal SMCsとnewborn rat SMCを用いて、PDGF-B鎖発現調節を検討した。 1)neointimal SMCおよびnewborn rat SMCでは、アンギオテンシンII(AngII)刺激によりPDGF-B鎖mRNAの発現が誘導された。これとは対照的に、adult rat SMCではPDGF-B鎖mRNAはAngIIによって誘導されなかった。また、AngII刺激によりnewborn rat SMCの培養上清中のPDGF-Bタンパクは増加した。 2)newborn rat SMCにおける、AngIIによるPDGF-B鎖mRNAの発現誘導は、AT1R拮抗薬CV11974で完全に抑制されたが、AT2R拮抗薬PD123319では抑制されなかった。 3)AngII刺激により3つのMAPキナーゼ(ERK,JNK,p38)のいずれも活性化された。MEK阻害剤であるPD98059は、AngIIによるPDGF-B鎖mRNAの発現をほぼ完全に抑制した。一方p38阻害剤であるSB203580には抑制作用はなかった。 4)Sis-Lucをトランスフェクトしたnewborn rat SMCをAngIIで刺激すると、ルシフェラーゼ活性が上昇した。また、Sis-Lucとともに各MAPキナーゼの優性抑制型発現ベクターを遺伝子導入した。AngIIによるルシフェラーゼ活性の増加は優性抑制型ERKおよび優性抑制型JNKの発現により抑制された。優性抑制型p38には抑制作用はなかった。 5)アデノウイルスを用いて優性抑制型Ras変異体を発現させると、AngIIによるERKの活性化とPDGF-B鎖mRNAの発現誘導は抑制された。しかしJNKの活性化は抑制されなかった。 6)サイクリックGMPおよびサイクリックAMPの誘導体により、AngIIによるERK及びJNKの活性化は抑制され、これとともにPDGF-B鎖mRNAの発現も抑制された。 以上、本論文は内因性PDGF-B鎖が内膜肥厚の形成期において重要な働きをしていることを始めて明らにした。さらに、PDGF-B鎖は内膜平滑筋細胞で産生され、アンギオテンシンIIによる調節を受けていることを示し、従来から重要と考えられた2つの増殖因子の直接の関連を示した。また本研究は、遺伝子導入法を用いて特定の増殖因子を選択的に阻害する方法を提示し、病態解明とともに臨床への応用が可能であり、学位に値するものと考えられる。 | |
| UTokyo Repositoryリンク | http://hdl.handle.net/2261/54720 |
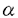 受容体のみと、B鎖はPDGF-
受容体のみと、B鎖はPDGF-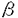 両受容体と結合し、受容体のチロシン残基の自己リン酸化をひきおこす結果、受容体を活性化する。特にPDGF-B鎖は血管平滑筋細胞に対し、強い増殖・遊走活性を有していることから、内膜肥厚の形成においてより重要な役割を果たしていることが示唆されてきた。実際、ラット頚動脈バルーン障害モデルを用い、PDGF-BBを持続注入すると内膜肥厚が増強されること、また、PDGF中和抗体を障害後7日間投与すると8日目の時点で内膜肥厚が40%抑制されることが報告されている。このほかに、私たちは、障害血管における受容体チロシンリン酸化レベルを測定することにより受容体の活性化を評価したところ、内膜肥厚が急速に進展する時期(障害後5日〜14日)に一致してPDGF-
両受容体と結合し、受容体のチロシン残基の自己リン酸化をひきおこす結果、受容体を活性化する。特にPDGF-B鎖は血管平滑筋細胞に対し、強い増殖・遊走活性を有していることから、内膜肥厚の形成においてより重要な役割を果たしていることが示唆されてきた。実際、ラット頚動脈バルーン障害モデルを用い、PDGF-BBを持続注入すると内膜肥厚が増強されること、また、PDGF中和抗体を障害後7日間投与すると8日目の時点で内膜肥厚が40%抑制されることが報告されている。このほかに、私たちは、障害血管における受容体チロシンリン酸化レベルを測定することにより受容体の活性化を評価したところ、内膜肥厚が急速に進展する時期(障害後5日〜14日)に一致してPDGF-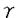 -32P]ATP存在下で各々ミエリン塩基性蛋白(ERK)、GST-c-Jun(JNK)、GST-ATF2(p38)のリン酸化を測定した.
-32P]ATP存在下で各々ミエリン塩基性蛋白(ERK)、GST-c-Jun(JNK)、GST-ATF2(p38)のリン酸化を測定した.