タンパク質は生体中においてその構造と機能を担う重要な生体物質である。これまでに、タンパク質工学は、目的のタンパク質をコードする遺伝子を天然酵素を用いて組み換えることによって多くの新規有用タンパク質を生み出してきた。しかし一方で、改変されたタンパク質が元のタンパク質と類似の立体構造をとって機能を発現するとは限らない。ゆえに、生理的条件下で立体構造を保持したままでタンパク質を直接に切断・再結合して新規有用タンパク質を生み出す技術は、次世代のバイオテクノロジーの有力な手法となることが期待できる。ところが、ペプチド結合は極めて安定な結合であり、生理的条件下での半減期はおよそ350年と見積もられている。したがって非酵素的にペプチド結合を加水分解するには、通常、強酸または強アルカリ存在下で長時間高温加熱する必要があり、生理的条件下で有効に作用する触媒の開発が強く求められている。これまでにペプチド結合を切断する触媒として,Co(III),Fe(III),Pd(II),およびCu(II)などの錯体が報告されているが、中性条件で高い活性を示すものはほとんどないのが現状である. 本研究では、希土類イオンおよび希土類-シクロデキストリン複合体が温和な条件でかつ効率的にペプチドを加水分解することを明らかにした。 第1章は序論である.本研究の背景にあるこれまでの経緯を述べ、さらに本研究を行う目的、意義について記述した。 第2章では、希土類イオンが、種々のペプチドを温和な条件で効率的に加水分解することを明らかに、さらに分光学、および動力学的に検討した。なかでも希土類イオンの1つであるCe(IV)はpH7.0、50℃の反応条件で、ジペプチドを半減期が数時間のオーダーで加水分解する。加速効果は10万倍以上である。ここで見い出された触媒活性はこれまでに報告された人工触媒のなかで最も高い. また同章において、反応機構の検討と高活性発現の要因について考察した。ペプチドの主鎖アミノ基とカルボキシル基、および切断されるアミド結合のカルボニル基がCe(IV)に配位し、カルボニル炭素を配位水酸基が求核攻撃して反応が進行する。その際、Ce(IV)は3価でも安定に存在できるため、高い電子受容性によりカルボニル基が効率的に活性化される。また、8〜9個の多数の配位座を有するので、基質と配位水酸基が立体的に有利な配向がとれる。さらに、ハードな希土類イオンは窒素より酸素との親和性が高いので、アミド結合の窒素原子でなくカルボニル酸素に配位する。そのために他の金属イオンで観測されるアミドの脱プロトンが中性条件でおこらない。このような要因のために、Ce(IV)は高い切断活性を生理的条件下で発現する。 さらにAsp側鎖のカルボキシル基とCe(IV)の相互作用を利用することにより、より効率的に加水分解が進行することを発見した。この知見に基づいて、タンパク質中のAspとGluの側鎖カルボキシル基がCe(IV)に配位することを利用して、タンパク質を位置選択的に加水分解できることを示唆した。 第3章では、Ce(IV)とシクロデキストリンの複合体が均一系でペプチドを加水分解することを示した。Ce(IV)は中性水溶液中で迅速に水酸化物の沈澱を形成し、系が不均一となる。これは反応機構の解明と将来の応用に重大な障害となっていた。このCe(IV)-シクロデキストリン複合体は、人工タンパク質分解酵素の活性部位に現在の時点で最も有望な金属触媒の1つであり、将来の大きな発展の可能性を持つ。 第4章では、種々の希土類イオンのアミノ酸エステルとアミノ酸アミドの加水分解活性を比較し、Ce(IV)のみがアミノ酸アミドを顕著に加速することを明らかにした。アミノ酸アミドの加水分解の律速段階は四面体中間体からのアミンの脱離であり、Ce(IV)がこの段階を酸触媒として効率的に加速する。ここで得られたCe(IV)の触媒作用についての基礎的知見は今後の触媒設計に大きな指針を与える。 第5章は総括である。ここまでの各章で述べた結果をまとめ、今後への展望を述べている。 以上のように本研究で示した希土類イオンのペプチド加水分解に対する有効性、ならびに反応機構の解明は,分子生物学、タンパク質工学の分野に大きく寄与し、他方面への工学的応用も期待できる。 よって本論文は博士(工学)の学位請求論文として合格と認められる。 | 




 -CH,
-CH, -CH2のピークが低磁場側にシフトした。続いてpDを5.7から6.8に上げると、さらにアミノ基側のGlyの
-CH2のピークが低磁場側にシフトした。続いてpDを5.7から6.8に上げると、さらにアミノ基側のGlyの

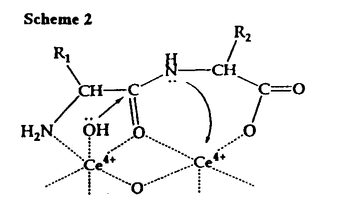
 -シクロデキストリン複合体によるジペプチドの加水分解
-シクロデキストリン複合体によるジペプチドの加水分解
