シリコンを基盤にした電子デバイス製造技術は既に成熟の域に達しており、次世代デバイスのための材料探索が行われだして久しい。しかし、化合物半導体を用いたデバイスが一部に存在するもののシリコンデバイスの圧倒的な優位性は変わっていない。近年の走査トンネル顕微鏡(STM)による原子スケールでのマニピュレーション技術の発展は、その応用において電子デバイスの飛躍的な集積化とその省電力化を期待させる。さらに、分子エレクトロニクスに代表されるように分子の自己組織化等を利用した新機能材料開発への展開が現実的になっている。また一方、理論的には第一原理計算の発展により様々な分子による表面再配列(吸着)構造およびその電子状態などが明らかにされている。しかしながら、吸着種(分子)間の相互作用はその重要性にも関わらず十分な理解が得られていない。本論文では、比較的小さなアセチレン(C2H2)分子とシリコン(Si)表面の吸着状態における相互作用を第一原理的手法を中心に用いて調べた。 低温(105K)におけるC2H2分子の吸着はキネティックアップテイク法を用いて調べられており、初期付着確率が1で、その飽和吸着表面は1Siダイマーあたりに1C2H2分子が吸着した状態(1 ML)であると結論されている。ところが最近、室温におけるSTM観察から飽和吸着表面は2Siダイマーあたり1C2H2分子が付着した状態(0.5 ML)であることが報告された。さらに、吸着の極めて初期段階においてもC2H2分子はその最近接サイトを互いに避けるように吸着する様子が観測されている。 従来の理論計算の報告では、この最近接サイトに吸着分子が存在しない特徴を最近接吸着分子間の反発相互作用による効果であるとしている。その第一原理計算の結果から見積もられた反発相互作用の大きさは190meVで、これは吸着による弾性歪みおよびSi-C結合で生じた電気双極子モーメント間の相互作用に起因し、特に前者が主要因であると報告されている。しかし、この理論計算は吸着分子のみに着目しており、Si(001)表面に存在するSiダイマーバックリングの取りうる二状態の自由度を無視している。そこで我々は、吸着分子数(表面被覆率)を変化させ、かつ二状態の自由度を考慮した第一原理計算を行い、その安定構造と全エネルギーを求めた。その結果、C2H2分子の吸着の有無を表す変数とSiダイマーのバックリングの二状態を表す変数から構成される二成分イジングモデルに、この計算結果をマッピングすることで全エネルギーを各相互作用部分に分解できることを見出した。 一方、熱力学的に安定な構造を決定するために、この系のグランドポテンシャルのC2H2分子の化学ポテンシャル依存性を調べたところ(図1参照)、安定構造は1ダイマーあたり1分子が吸着した構造であることが分かった。すなわち、十分な分子の表面への暴露と十分な時間を経過すればC2H2分子は全てのSiダイマー上に吸着することを表している。この結果は、低温におけるキネティックアップテイク法の実験結果と矛盾しない。また、この図からSi(001)表面に対するC2H2分子の吸着が非常に局在した現象であることが読み取れる。 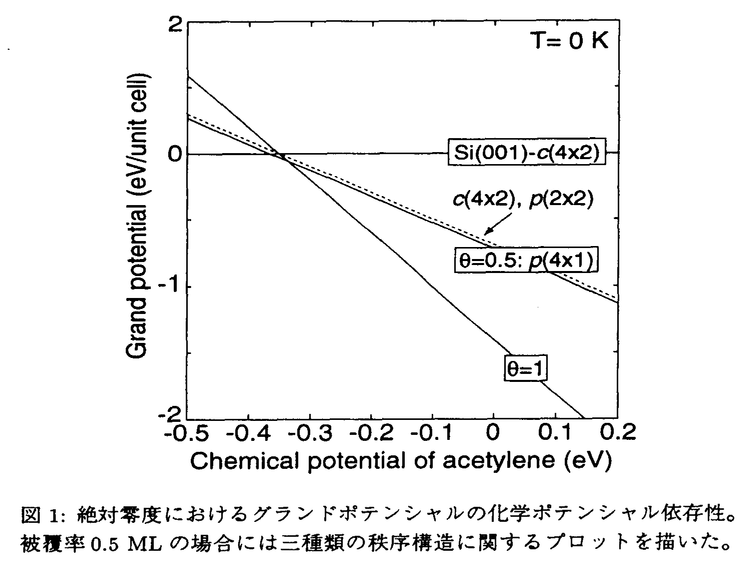 図1:絶対零度におけるグランドポテンシャルの化学ポテンシャル依存性。被覆率0.5MLの場合には三種類の秩序構造に関するプロットを描いた。 図1:絶対零度におけるグランドポテンシャルの化学ポテンシャル依存性。被覆率0.5MLの場合には三種類の秩序構造に関するプロットを描いた。 室温STM観察によれば、最近接サイトに吸着分子は存在しない。ところが、第一原理計算による被覆率0.5MLにおける最も安定な吸着構造はp(4×1)構造である。この構造では吸着分子がSiダイマー列に沿って全て吸着した列および表面Siダイマーが列に沿って交互にバックリングした列の両者が交互に存在している。計算結果からマッピングされた吸着分子間の最近接相互作用の値は24.8meVの弱い反発相互作用である。しかし、残った表面Siダイマーがその向きを列に沿って交互に整列する場合のエネルギーの得分が上回るためにp(4×1)構造が安定となる。このことは最近接サイトに吸着分子が存在しない理由が吸着現象の動的過程によるものであることを示唆している。また、従来の報告とは異なり、吸着分子間の最近接相互作用は吸着によって生じる電気双極子モーメント間の相互作用が主な要因であり、弾性歪みの寄与は比較的少ないことが分かった。 この系は、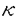 BTよりも十分大きな吸着エネルギーを持つために通常の観測時間では熱平衡状態に達しないものと考えられる。しかしながら、被覆率0.5MLにおける各配列構造のエネルギー(図2参照)から、熱平衡状態を仮定した場合の有限温度での振舞いは興味深いものと考えられる。そこで、吸着エネルギー項を除いた仮想的なモデルハミルトニアンを用いて有限温度でのSiダイマーの取りうる二状態によるエントロピー効果を調べた。 BTよりも十分大きな吸着エネルギーを持つために通常の観測時間では熱平衡状態に達しないものと考えられる。しかしながら、被覆率0.5MLにおける各配列構造のエネルギー(図2参照)から、熱平衡状態を仮定した場合の有限温度での振舞いは興味深いものと考えられる。そこで、吸着エネルギー項を除いた仮想的なモデルハミルトニアンを用いて有限温度でのSiダイマーの取りうる二状態によるエントロピー効果を調べた。  図2:被覆率0.5MLにおける各配列構造のエネルギーダイアグラム(数値の単位はmeV)。Siダイマーの向きで隔てられたエネルギー障壁はダイマーが水平の場合のエネルギーである。 図2:被覆率0.5MLにおける各配列構造のエネルギーダイアグラム(数値の単位はmeV)。Siダイマーの向きで隔てられたエネルギー障壁はダイマーが水平の場合のエネルギーである。 吸着位置を固定した配列構造に対する自由エネルギーは熱力学的極限での厳密解から450Kでp(2×2)構造がp(4×1)構造よりも小さくなることが分かった。さらに、拘束のない配列構造および表面欠陥の導入された構造に対してメトロポリス法によるモンテカルロシミュレーションを用いて調べた。その結果、最近接間の相関関数が450K付近で減少すること、表面欠陥の増加によってこの現象が強調されることが分かった(図3参照)。これは、Siダイマーの二状態によるエントロピー項への寄与が比較的低温から最近接サイト間の相関関数に影響を及ぼすことを表している。また、表面欠陥による強調効果は、Siダイマー間相互作用の異方性、すなわち最近接間相互作用が第二近接以降よりもかなり大きなことの反映と考えられる。  図3:被覆率0.5MLにおける吸着位置固定での各配列構造に対する自由エネルギーの厳密解の温度変化(左下図)とモンテカルロシミュレーションによる相関関数の温度依存性(右図)。図中の1、2A、3はそれぞれ左上図(Mは吸着サイト)の位置に対応する。(a)表面欠陥のない場合、(b)表面欠陥を5%含む場合、(c)表面欠陥を20%含む場合。 図3:被覆率0.5MLにおける吸着位置固定での各配列構造に対する自由エネルギーの厳密解の温度変化(左下図)とモンテカルロシミュレーションによる相関関数の温度依存性(右図)。図中の1、2A、3はそれぞれ左上図(Mは吸着サイト)の位置に対応する。(a)表面欠陥のない場合、(b)表面欠陥を5%含む場合、(c)表面欠陥を20%含む場合。 |