| 内容要旨 | | 有機亜鉛試薬を用いた不斉アルキル化反応のいくつかはほぼ完全に立体制御された生成物を与えるまでに開発が進んでいる.ところが有機金属試薬や基質の溶液内での会合状態や遷移状態の詳細な情報は明らかになっていない部分が多い. アルデヒドの不斉アルキル化に用いられている光学活性アミノアルコール-ジアルキル亜鉛試薬は2量体であるといわれており,不斉誘起の機構が複雑であると予想される.本研究にて発表者は,会合状態を制御した光学活性アリル亜鉛試薬を用いた炭素-ヘテロ原子2重結合へのエナンチオ選択的アリル化反応について実験的検討を行い,高い選択性をもって付加体を得ることに成功した. また,RZnX型の有機亜鉛試薬は溶液中で2量体以上で存在することが多く,かつ溶媒によって会合状態が異なることが予想される.ところが,会合体の反応に及ぼす影響を実験的手法で調べるのは困難である.発表者は理論的手法を用いてアリル亜鉛試薬のビニル金属種への付加反応やSimmons-Smith反応について検討し,有機亜鉛試薬の会合状態と反応性との関係に関する重要な知見を得た. アルキニルケトンの不斉アリル化反応 発表者は修士課程での研究において,ビスオキサゾリン型不斉配位子を有する光学活性アリル亜鉛試薬の種々のイミンへの付加反応を検討した.その結果,3,4-ジヒドロイソキノリンのような環状Z-イミンへの付加反応ではアリル化生成物が最高99% eeで得られた(式1).本光学活性アリル亜鉛試薬は亜鉛原子の配位様式から単量体で存在していることが示唆され,また,本反応は強固な6中心遷移状態を経て進行していることが示唆された. 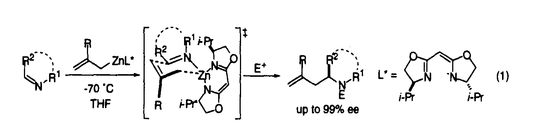 一方,アルデヒドの不斉アルキル化反応と比べ,ケトンの不斉アルキル化反応は一般に困難である.その理由は双方の置換基がアルキル基であることから置換基の嵩高さに大きな差がないためと考えられる.発表者は,ケトンの一方の置換基に水素原子程度の大きさと考えられているC-C三重結合を有する , , -アルキニルケトンを基質として検討を行った. -アルキニルケトンを基質として検討を行った. リチオ化した不斉配位子L*と臭化アリル亜鉛とから調製した1を,-アルキニルケトン2に低温にて作用させたところ,対応する3級アルコール3を得た(式2).1を用いたアルデヒドの不斉アリル化反応では選択性が低いものの,2の不斉アリル化反応では高いエナンチオ選択性をもって生成物を与えた.また,基質上の置換基について検討したところ,アルキニル基上の置換基R1は小さい方が選択性が高く(Table 1.entries 1-3),R2は逆に嵩高いほうが選択性が高いことがわかった(entries 3-5).  生成物3の絶対配置と分子軌道計算の結果から,本反応はアルキル基R2がエカトリアル位にあり,かつアキシアル位のアルキニル基と不斉配位子上の置換基との立体反発が最小となるような配座の遷移構造をへて進行していることが示唆された(Figure 1).  図表Table 1.Enantioselective allylation of ,-ynones / Figure 1.The most favorable transition structure proposed on the basis of the ab initio calculationsSimmons-Smith反応に関する理論的検討 図表Table 1.Enantioselective allylation of ,-ynones / Figure 1.The most favorable transition structure proposed on the basis of the ab initio calculationsSimmons-Smith反応に関する理論的検討 光学活性ルイス酸存在下でのアリルアルコールのSimmons-Smith(SS)反応は低温で速やかに進行し光学活性シクロプロピルメタノールを高い不斉収率で与える.ところが,SS反応のルイス酸による反応加速の機構については余り理解が進んでいない.すなわちアリルアルコールへのSS反応では,ルイス酸がSS試薬の亜鉛原子上の配位子であるアルコキシ酸素に配位し,間接的にハロゲン化物イオンの脱離を促進する機構(mode A)が専ら提唱されている.ところが,オレフィンへのSS反応では,ルイス酸がSS試薬のハロメチル部位に配位し,ハロゲン化物イオンの脱離を直接促進する機構(mode B)も提案されたこともある.発表者はアリルアルコールのSS反応におけるルイス酸の働きと,有機亜鉛試薬会合体が反応に及ぼす効果について興味をもち,密度汎関数法計算(B3LYP/631A level)を行った. 当初,1分子のアリルアルコールがSS試薬によって脱プロトン化されたクロロメチル亜鉛アリロキシドをモデルとしたが,このモデルのSS反応の遷移構造TS2の活性化エネルギーは35.7 kcal/molと非常に高く,妥当なモデルではないことが示唆された.そこで,亜鉛アルコキシドは結晶構造中ではオリゴマーとして存在することが知られていることから,基質モデルを亜鉛アリロキシド2量体とした.モデルの簡略化のため,亜鉛アリロキシド2量体中の反応に関与しないアルキル基は全てメチル基に置き換えて検討した. まずmode Aの錯体モデルCP2はルイス酸がアルコキシ酸素上に配位するモデルであり,これまで頻繁に提唱されているモデルである(Figure 3).ところが,遷移構造TS3の活性化エネルギーは27.9kcal/molと高く,ルイス酸による反応活性化の効果はほとんどないことが明らかとなった.理由はこの亜鉛原子にはアルコキシ酸素2原子がすでに配位しているため,塩素原子の配位が弱いためと考えられる.  Figure 3.Mode A reaction pathway of the cyclopropanation of allylalkoxide dimer Figure 3.Mode A reaction pathway of the cyclopropanation of allylalkoxide dimer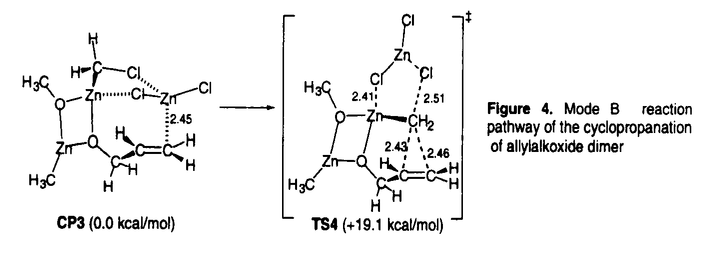 Figure 4.Mode B reaction pathway of the cyclopropanation of allylalkoxide dimer Figure 4.Mode B reaction pathway of the cyclopropanation of allylalkoxide dimer 一方,mode Bの錯体モデルCP3においてはルイス酸がハロメチル塩素に配位した5中心の構造が得られた.遷移構造TS4の活性化エネルギーは19.1kcal/molと大幅に低下し,かつTS4はTS3よりも5.2kcal/mol安定化した.アリルアルコールへのSS反応はmode Bを経て進行していることが示唆された. また亜鉛アルコキシドオリゴマーを形成すると有利になる理由は,活性錯体の反応部位同士が接近した立体配座をとるためと考えられる. アリル亜鉛試薬のビニル金属種への付加反応の理論的検討 生成物としてgem-二核金属種を与えるアリル亜鉛試薬のビニル金属種への付加反応は,形式的にはアニオンのアニオンへの求核攻撃であり,本来非常に不利な反応である.本反応の反応機構としてマグネシウム塩が関与したアリルビニル亜鉛のメタラクライゼン転位をへて進行することが提案されているが,亜鉛・マグネシウムの2種の金属原子がどのように反応に関与しているかは不明であった.発表者は塩化アリル亜鉛と塩化ビニルマグネシウムの反応をモデルとして密度汎関数法計算(B3LYP/631A level)を行い,本反応の反応経路の詳細を検討した(Figure 2). 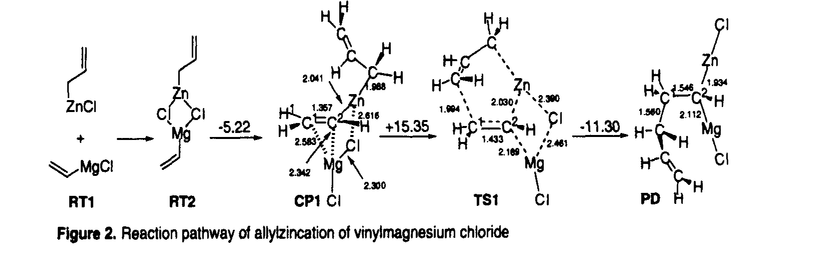 Figure 2.Reaction pathway of allylzincation of vinylmagnesium chloride Figure 2.Reaction pathway of allylzincation of vinylmagnesium chloride 本反応条件下では,RMX型の有機金属試薬は2量体以上のオリゴマーで存在することが予想される.出発物質RT1から錯体化・トランスメタル化を経てクラスター構造CP1を形成し,その構造を保持しつつ反応が進行する経路について検討を行った.その結果,本反応はルイス酸によって活性化された亜鉛上でのメタラクライゼン転位と呼びうる遷移状態TS1を経ることが示唆された.一方,生成物の二核金属化合物PDはCP1よりも不安定であるものの,トランスメタル化を経たジアルキル亜鉛オリゴマーの形成により安定化されることが示唆された.本反応系では,生成物の安定化のため炭素-金属結合が安定な亜鉛と,遷移状態の活性化エネルギーの低下のためにマグネシウムなどのルイス酸性の強い金属の存在が必須であることが明らかとなった. さらに,1-propenylmagnesium chlorideとcrotylzinc chlorideを用いたジアステレオ選択性の問題が生じる系の検討は実験結果をよく再現し,本モデルの妥当性を支持している. |
| 審査要旨 | | 本論文は5章からなり,第1章は序論で,有機亜鉛試薬を用いた立体選択的反応の現状について述べられている.第2章から4章は論文提出者の研究結果で,第2章はアリル亜鉛試薬のアルキニルケトンへのニナンチオ選択的付加反応,第3章はSimmons-Smith反応の理論的検討,第4章はアリル亜鉛試薬のビニル金属種への付加反応の理論的検討についてそれぞれ述べられている.第5章はそれらの結果のまとめと今後の展望について述べられている. 第2章:アルデヒドの不斉アルキル化反応と比べ,ケトンの不斉アルキル化反応は一般に困難である.その理由は双方の置換基がアルキル基であることから置換基の嵩高さに大きな差がないためと考えられる.論文提出者は,ケトンの一方の置換基に水素原子程度の大きさと考えられているC-C三重結合を有する,-アルキニルケトンを基質として検討を行った. アニオン型のビスオキサゾリン型不斉配位子を有する光学活性アリル亜鉛試薬を,-アルキニルケトンに低温にて作用させたところ,高いエナンチオ選択性をもって対応する3級アルコールを得ることに成功した.得られた生成物の絶対配置と分子軌道計算の結果から,本反応は6中心遷移状態において,アルキル基R2がエカトリアル位にあり,かつアキシアル位のアルキニル基と不斉配位子上の置換基との立体反発が最小となるような配座の遷移構造をへて進行していることが示唆された. 第3章:Simmons-Smith(SS)反応はルイス酸によって加速されることが知られているものの,その反応加速の機構については余り理解が進んでいない.すなわちアリルアルコールへのSS反応では,ルイス酸がSS試薬の亜鉛原子上の配位子であるアルコキシ酸素に配位し,間接的にハロゲン化物イオンの脱離を促進する機構(mode A)が専ら提唱されている.ところが,オレフィンへのSS反応では,ルイス酸がSS試薬のハロメチル部位に配位し,ハロゲン化物イオンの脱離を直接促進する機構(mode B)も提案されたこともある.論文提出者はアリルアルコールのSS反応におけるルイス酸の働きについて興味をもち,密度汎関数法計算を行った. 当初,1分子のアリルアルコールがSS試薬によって脱プロトン化されたクロロメチル亜鉛アリロキシドをモデルとしたが,遷移構造の活性化エネルギーの高さから(35.7 kcal/mol)妥当なモデルではないことが示唆された.そこで,亜鉛アルコキシドは結晶構造中ではオリゴマーとして存在することが知られていることから,基質モデルを亜鉛アリロキシド2量体として検討した. まずmode Aのモデルの遷移構造の活性化エネルギーは27.9kcal/molと高く,ルイス酸による反応活性化の効果はほとんどないことが明らかとなった.理由はこの亜鉛原子にはアルコキシ酸素2原子がすでに配位しているため,塩素原子の配位が弱いためと考えられる.一方,mode Bのモデルの遷移構造の活性化エネルギーは19.1kcal/molと大幅に低下し,アリルアルコールへのSS反応はmode Bを経て進行していることが示唆された. 第4章:生成物としてgem-二核金属種を与えるアリル亜鉛試薬のビニル金属種への付加反応は,形式的にはアニオンのアニオンへの求核攻撃であり,本来非常に不利な反応である.本反応の反応機構としてマグネシウム塩が関与したアリルビニル亜鉛のメタラクライゼン転位をへて進行することが提案されているが,亜鉛・マグネシウムの2種の金属原子がどのように反応に関与しているかは不明であった.論文提出者は塩化アリル亜鉛と塩化ビニルマグネシウムの反応をモデルとして密度汎関数法計算を行った. 本反応条件下では,RMX型の有機金属試薬は2量体以上のオリゴマーで存在することが予想される.論文提出者は出発物質から錯体化・トランスメタル化を経てクラスター構造の錯体を形成し,その構造を保持しつつ反応が進行する経路について検討を行った.その結果,本反応はルイス酸によって活性化された亜鉛上でのメタラクライゼン転位と呼びうる遷移状態を経ることが示唆された.一方,生成物の二核金属化合物はクラスター構造の錯体よりも不安定であるものの,トランスメタル化を経たジアルキル亜鉛オリゴマーの形成により安定化されることが示唆された.本反応系では,生成物の安定化のため炭素-金属結合が安定な亜鉛と,遷移状態の活性化エネルギーの低下のためにマグネシウムなどのルイス酸性の強い金属の存在が必須であることが明らかとなった. なお,第2章は,中村正治,曽木美希との共同研究,第3,4章は中村正治との共同研究であるが,論文提出者が主体となって分析および検証を行ったもので,論文提出者の寄与が充分であると判断する. 従って,博士(理学)の学位を授与できるものと認める. |