老人斑と神経原線維変化はアルツハイマー病(Alzheimer’s disease,AD)の脳に出現する特徴的な異常構造物である。老人斑はアミロイド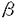 蛋白(A)の沈着がその本態であり、AD発症の原因と考えている研究者も多い。しかし、老人斑の出現数と痴呆の程度に相関はなく、Aの脳内沈着をきたすトランスジェニックマウスに神経原線維変化も神経細胞死も生じないことから、A原因説を疑問視する根強い声があることも事実である。一方、神経原線維変化は高度にリン酸化したtauが重合したpaired helical filament(PHF)から出来ている。その出現頻度は痴呆や脳に認められる神経細胞死の程度と良く相関することから、ADの病態メカニズムの発生に深く関与していることが疑われるが、AD以外の神経疾患にも見られることから、2次的な変化である可能性が否定できなかった。昨年、アルツハイマー病と同じくtauの蓄積がみられる家族性前頭側頭型痴呆(Frontotempolal dementia and parkinsonism linked to chromosome 17:FTDP-17)とよばれる一群の痴呆疾患の原因遺伝子がtauであることが明らかとなった。この発見はtau自体の異常が神経原線維変化と神経細胞死の必要十分条件であることを初めて証明したものであり、FTDP-17を1次性タウオパチーとすれば、ADを2次性タウオパチーとしてとらえる可能性が出てきたことを意味している。すなわち、FTDP-17の発症メカニズムを解明することによって、ADをはじめとした他のタウオパチーの発症メカニズムの解明への糸口が見つかる可能性が出てきたわけである。 蛋白(A)の沈着がその本態であり、AD発症の原因と考えている研究者も多い。しかし、老人斑の出現数と痴呆の程度に相関はなく、Aの脳内沈着をきたすトランスジェニックマウスに神経原線維変化も神経細胞死も生じないことから、A原因説を疑問視する根強い声があることも事実である。一方、神経原線維変化は高度にリン酸化したtauが重合したpaired helical filament(PHF)から出来ている。その出現頻度は痴呆や脳に認められる神経細胞死の程度と良く相関することから、ADの病態メカニズムの発生に深く関与していることが疑われるが、AD以外の神経疾患にも見られることから、2次的な変化である可能性が否定できなかった。昨年、アルツハイマー病と同じくtauの蓄積がみられる家族性前頭側頭型痴呆(Frontotempolal dementia and parkinsonism linked to chromosome 17:FTDP-17)とよばれる一群の痴呆疾患の原因遺伝子がtauであることが明らかとなった。この発見はtau自体の異常が神経原線維変化と神経細胞死の必要十分条件であることを初めて証明したものであり、FTDP-17を1次性タウオパチーとすれば、ADを2次性タウオパチーとしてとらえる可能性が出てきたことを意味している。すなわち、FTDP-17の発症メカニズムを解明することによって、ADをはじめとした他のタウオパチーの発症メカニズムの解明への糸口が見つかる可能性が出てきたわけである。 昨年FTDP-17の原因遺伝子がtauであることが判明してから、いくつかのグループがcell-free系を用いて変異型tauの特性を報告している。試験管内でチューブリンとtauを混合し、その微小管重合促進活性を調べると、FTDP-17にみられる変異型tauでは微小管の重合促進活性が正常型tauよりも低下しているということが明かとなった。しかし、(1)同様の実験であっても研究グループによって結果が異なること、(2)実験系への他のタンパク質や核酸の混入は異なった結果を導くこと、すなわち純粋にtauとチューブリンだけが存在する条件下でなければ実験が成立しないこと、(3)実験に用いたtauはバクテリアで合成されているのでリン酸化されていないのでコンフォメーションが異なる、などの理由からcell-freeの実験結果が生体内での現象を反映しているとは言い難いと思われる。この点、培養細胞系は多くの利点をもつが、他の微小管結合タンパク質と機能を代償しあってしまう可能性や、発現する細胞や分化の程度によって結果が異なってくる可能性がある。そこで我々は、FTDP-17で発見された4つのtau変異株(G272V、P301L、V337M、R406W)と野生型tauを安定的に発現するCHO細胞株を構築し、生化学的、免疫細胞化学的解析を行った。 まず始めに、これらの変異がtauの微小管結合領域とその近傍に存在することから、tauの微小管への結合能について解析した。通常の培養条件では、tauの細胞内分布と微小管との結合パターンに野生株と変異株との間で相違は見られなかった。ところが、微小管脱重合試薬のコルセミドで処理した場合、G272VとP301L株は野生株を含む他の細胞株と比較して微小管が脱重合しやすく、変異tauが微小管の安定化に影響を与えていることが示唆された。 最近、tauを細胞に強制発現させるとキネシン依存性のオルガネラ輸送に影響をおよぼし、ミトコンドリアや小胞体の細胞内輸送や分布に支障をきたすという報告がされた。そこで、ミトコンドリアとリソソームの細胞内局在について調べた。tauを強制発現させない場合、ミトコンドリアとリソソームは核周囲と細胞質内にかなり広範に存在していた。ところが、野生型tauを強制発現させるとミトコンドリアは核の周囲に集積し、細胞の辺縁には認められなくなった。変異型tauについて検討したが、野生型tau発現株と比べてミトコンドリアの分布に違いは見られなかった。一方、リソソームの分布は野生型tauの発現やtau変異の影響を受けなかった。 培養細胞の細胞膜直下のアクチンフィラメントのネットワークをサイトカラシンによって破壊すると細胞が突起を延ばすようになる。この突起の伸長過程には微小管の重合が関与していることが知られている。そこで微小管の重合を外的に誘導した場合に、野生型と変異型との間でtauの微小管重合促進能に違いがあるかどうかを調べた。その結果、tauを発現させない場合が最も突起の伸長が著しく、tauを発現させた場合突起の伸長は遅延することがわかった。しかし野生型と変異型との間では差は見られなかった。 次にtauのリン酸化を調べた。ウエスタンブロットにより野生型tauを検出すると、リン酸化の相違によって移動度のことなる特徴的な3つのバンドが現れた。このことはCHO細胞で発現させたtauは内在性のキナーゼにより、リン酸化されていることを示唆している。tau1抗体で染色すると、野生型を含め多くの株ではリン酸化されている最も上部のバンドが一番濃く、脱リン酸化された移動度の最も早いバンドは薄く認識された。興味深いことに、R406W株ではこのバンドの認識パターンが全く逆だった。これはR406W株だけはリン酸化のパターンが異なっていることを意味している。そこでさらに詳しく、R406W株のリン酸化について検討した。406番目に最も近いリン酸化部位であるSer396とSer404のリン酸化を特異的に認識する抗体であるPHF1で染色すると、R406W株では野生株や他の変異株に比べて反応性が低下していた。さらにSer396のリン酸化だけを認識する特異抗体(C5)で検討すると、R406W株での同部位のリン酸化は著しく低下していた。したがって、406番目のアルギニンがトリプトファンに置換されることにより396番目のセリン残基がリン酸化されにくくなったと考えられる。さらに、他のリン酸化部位を認識する抗体の反応性をみたが、R406W株ではThr231のリン酸化も低下していることが明らがとなった。しかし、実際にtau遺伝子にR406Wの変異を持つ患者の脳ではPHF様の構造物が蓄積し、Ser396・Thr231は過剰にリン酸化されている。したがって患者の脳内ではリン酸化の程度の低いSer396やThr231を過剰にリン酸化させる状態が生じていることが予想され、これはR406W株で過リン酸化が生じる条件と類似しているはずである。 そこで、R406W株においてtauのSer396がリン酸化されるような条件を検討すべく、オカダ酸によるホスファターゼ活性の阻害を試みた。無処理の際、R406W株ではSer396のリン酸化が弱かった。ところが、オカダ酸処理によりR406W株でも他の株と同様にSer396のリン酸化が強くなった。これは、オカダ酸によりPP2A(protein phosphatase 2A)およびPP1(protein phosphatase 1)が阻害され、相対的にキナーゼ活性が強くなりSer396がリン酸化されたと考えられる。 さらに、各細胞周期におけるtauのリン酸化について調べた。G1期とS期の間、あるいはS期で細胞周期を停止させた場合、R406W株は、コントロールと同様Ser396のリン酸化が微弱であった。ところが細胞周期をノコダゾールでM期に停止させると、Ser396のリン酸化が野生株と同等に強くなった。以上の結果から、R406W tauであっても細胞分裂のような特別な条件であればSer396がリン酸化されることが示唆された。 さらに、細胞周期をM期で停止させると、AD脳で高度にリン酸化されたtauを認識するAP422抗体やAT8抗体のシグナルが野生型およびR406W tauで現れた。AP422抗体はSer422のリン酸化を認識する抗体である。AP422抗体はAD以外の種々の神経変性疾患(ダウン症候群、脳炎後パーキンソニズム、進行性核上性麻痺、皮質基底核変性症、ピック病等)の繊維性沈着物にも反応性があることが報告されている。したがって、tauのSer422は多くの神経変性疾患に共通して過剰なリン酸化が起こる場所であり、M期におけるtauのリン酸化状態と非常に類似するといえるであろう。 成人の神経細胞は一般的に細胞分裂はせず、極めて分化した状態にあると考えられている。ところがAD脳の神経細胞は必ずしも静止した状態(Go)ではなく、細胞周期が進行しているような表現形を示すことが知られている。たとえばAD脳では細胞分裂に必要なcdc2やサイクリンB1の抗体反応性がある。AD脳で発現しているcdc2は活性型であることが活性型cdc2特異的抗体により確認されている。このようなcdc2やサイクリンB1の再発現は、正常老化脳や成熟マウス脳では見られずAD脳の神経細胞で見られる。さらに、細胞の生育を促すcdk2やcdk4がAD脳内の神経細胞に存在することから、これらの細胞はもはや静止した状態ではないことを示している。しかしながら核の分裂や染色体の凝集などがみられない。 本研究では、未分化Neuro2a細胞における薬剤に対する細胞毒性についても検討したが、tauの発現による感受性の変化は認められなかった。おそらく上記のようなAD脳における環境を再現できなかったためと推察される。本研究で用いたCHO細胞の系は神経系の細胞ではないが、生体内での病態の一部を再現する。  FTDP-17におけるtauのミスセンス異変と本研究に用いた抗体 FTDP-17におけるtauのミスセンス異変と本研究に用いた抗体 |