CP-225,917(1)およびCP-263,114(2)は、近年Pfizer社の研究者により未確認の菌類から単離された。1および2はスクアレン合成酵素およびファルネシル基転移酵素に対して阻害活性を示すことが報告されている。これらの化合物はこうした興味深い生理活性のほか、橋頭位二重結合、ラクトン-アセタールまたは-ヘミアセタール構造、および無水マレイン酸を部分構造として含む、複雑かつ特徴的な多環性の骨格を有している。構造決定は主として各種NMRスペクトルの詳細な検討によって行われ、立体化学に関しては相対配置のみが知られている。最近、Nicolaouらにより1および2のラセミ体の全合成が達成されているが、光学活性体の合成は未だ達成されていない。また、Danishefskyをはじめ、多くの合成化学者により活発に合成研究が行われている。筆者は分子内Diels-Alder反応を骨格構築における鍵反応として用い、2の光学活性体の全合成を達成すべく検討を行ったので、以下その概略を述べる。  Figure 1.Structures of CP-225,917(1)and CP-263,114(2). Figure 1.Structures of CP-225,917(1)and CP-263,114(2). CP-263,114の逆合成解析をScheme1に示す。まず、光学活性な側鎖を有するAを合成中間体として考え、骨格を分子内Diels-Alder反応により構築することにした。その前駆体はフラグメントB、Cとそれらを連結させるアクリル酸誘導体へと分割可能と考えた。フラグメントBについては適当な光学活性な化合物から合成することとした。一方、分子内Diels-Alder反応を円滑に進行させるには1,3-ジエン部位がs-cisの配座をとりうることが必須である。したがってフラグメントCはScheme1に示した幾何配置のものを合成しなければならない。そのための手法として、末端に置換基を有するアレンカルボン酸エステルに対するアルケニル金属種の1,4-付加反応を用いることとした。  Scheme 1 Scheme 1 まず、この合成戦略の有効性を検証するためにラセミ体でモデル検討を行った。アレンカルボン酸エステル4は、容易に合成可能な3の異性化反応により調製した。典型的な銅試薬であるジアルケニル銅リチウム5と4の反応では目的とする付加体7の他に8を与えた(Table 1,Entry 1)。そこで検討したところ、THF-Me2S混合溶媒中、HMPAおよびTMSClを添加剤として用いることにより望みとする7を収率良く単一の幾何異性体として得ることができた(Entry 2)。この条件ではアルケニル銅6を1当量用いるだけで良好な結果が得られており、本反応は実際の合成にも適用可能と期待できる。  Table 1.1,4-Addition of Alkenyl Group to Allenecarboxylate 4. Table 1.1,4-Addition of Alkenyl Group to Allenecarboxylate 4. こうして得られた7を4工程の変換を経て分子内Diels-Alder反応前駆体9とした(Scheme 2)。このものにルイス酸として二塩化エチルアルミニウムを作用させたところ分子内Diels-Alder反応は円滑に進行し、二環性化合物10を単一のジアステレオマーとして得ることができた。なお、この反応において少量のピリジンの添加はエチルチオ基に関する幾何異性体の生成といった副反応の抑制に必須であった。また、10はX線結晶解析により望みとする相対配置であることを確認した(Figure 2)。こうして、分子内Diels-Alder反応が2の基本骨格構築に有効なことを示した。さらに、得られた10をジケトン11へと変換することに成功し、骨格上の官能基変換も可能であることを確認した。  図表Scheme 2 / Figure 2.ORTEP drawing of 10. 図表Scheme 2 / Figure 2.ORTEP drawing of 10. 次に、光学活性体の合成について検討を行った。そのための手法としては、Evansらにより開発されたキラル補助基を用いるアルドール反応を適用することとした(Scheme 3)。先述の1,3-ジエン7を12へと変換した後、ホウ素エノラートを用いるアルドール反応を行ったところ、付加体13を単一のジアステレオマーとして得ることができた。そして、水酸基の酸化を行った後に塩化亜鉛-ジエチルエーテル錯体を少量のピリジン存在下に作用させたところ、先のモデル基質と同様に分子内Diels-Alder反応が進行し、望みとする環化体14を単一のジアステレオマーとして得ることに成功した。  Scheme 3 Scheme 3 こうして光学活性な基本骨格の構築が達成できたので、骨格上の官能基変換について検討を行った(Scheme 4)。まず環化体14のキラル補助基を除去して15とした。このものをエノールトリフラート16とした後にPummerer転位によるカルボニル基の導入を行い、17を得た。このものはPMB基の除去の後、容易にラクトンアセタール18を与えた。しかしながら、カルボニル挿入反応によるマレイン酸部位の構築は達成できなかった。一方、ラクトンアセタールの形成以前の17を用いればカルボニル挿入反応は進行し、満足すべき収率ではないものの望みとする19を得ることができた。そして、ラクトン-アセタールの形成を行い、20の合成に成功した。ところが、このラクトン部位は容易に求核攻撃を受けてマロン酸エステル部位の開裂を起こし、21を与えることが判明した。そこでその後の変換を断念し、このようなラクトン化によらずにエステルを区別する方法を検討することにした。  Scheme 4 Scheme 4 検討した結果、15を水酸化バリウムにより加水分解すると選択的にジカルボン酸22を与えることを見いだした(Scheme 5)。さらに、このものをクロロギ酸メチルおよびトリエチルアミンを用いて選択的にエステル化するとモノカルボン酸23を生成することが判明した。こうしてエステル基の区別が可能であることが明らかとなったので、光学活性な側鎖を用いた2の合成に着手した。 モデル基質12と同様にして合成した、天然物と同じ長鎖アルキル基を有する25と、容易に入手可能な天然型の(S)-リンゴ酸から誘導した , , -不飽和アルデヒド24とのアルドール反応により付加体26を合成した。さらに水酸基の酸化および分子内Diels-Alder反応により基本骨格を構築し、27を得た(Scheme 6)。 -不飽和アルデヒド24とのアルドール反応により付加体26を合成した。さらに水酸基の酸化および分子内Diels-Alder反応により基本骨格を構築し、27を得た(Scheme 6)。 これをScheme 5の方法によりモノカルボン酸28とした後にArndt-Eistert法による一炭素増炭を試みた(Scheme 7)。ジアゾケトン29をメタノール中で安息香酸銀とともに加熱したところ30を低収率で与えた。収率が低い理由として、カルベンのケトエステル部位に対するC-H挿入反応を経由し、反応系が複雑化したと考えた。そこで、11,12位間が二重結合となった31を合成して先程と同様に銀塩で処理したところ望みとする転位反応が進行し、32を得ることができた。しかしながら、29からのエノールトリフラートの形成は再現性に問題があり、無水マレイン酸部位の構築について新たな手法の開発が必要となった。 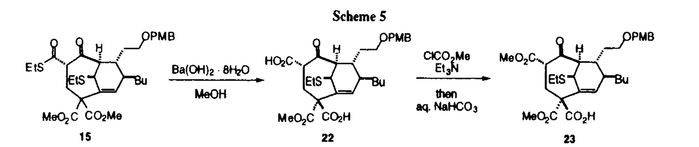 Scheme 5 Scheme 5 Scheme 6 Scheme 6 Scheme 7 Scheme 7 種々検討したところ、天然物の全合成に必要な全ての官能基を有する化合物36を得ることに成功した。マレイン酸部位の構築については分子内アルドール反応および脱炭酸を利用する手法を見いだした。(Scheme 8)。すなわち、27に対してメルカプト酢酸アリルのリチウム塩を作用させてキラル補助基を除去し、さらにDBUで処理することにより分子内アルドール反応を行って33を得た。そして、このものをアリル基の除去の後、ピリジン存在下に無水酢酸中で加熱することにより脱水と脱炭酸が進行し、チオブテノライド34へと変換することができた。チオブテノライドの酸化によって無水マレイン酸部位を構築した後にエステルの区別とAmdt-Eistert法による一炭素増炭を行い、35を得た。さらに、Pummerer転位とラクトン-アセタールの形成を行い、36を得ることができた。そして、36について天然物と同様のNOEが観測されたことから、天然物と同じ相対配置であることが確認できた。  Scheme 8 Scheme 8 ここで、より収束性の高い合成ルートの開発を目指し、光学活性な側鎖部分の合成法の改良を行った(Scheme 9)。基質としては(S)-エピクロロヒドリンを用いた。このものはJacobsenらにより開発された速度論的分割法を用いて容易に入手可能である。アセチリドによるオキシラン環の開裂を経て37とし、このもののPummerer転位により生じたアルデヒドに対するGrignard試薬の付加を経て、光学活性な側鎖部分に必要な全ての炭素原子を有する,-不飽和アルデヒド38を得ることができた。  Scheme 9 Scheme 9 このものを用いてScheme6と同様に骨格の構築を行い、40を得た。さらにScheme 8に示した手法により無水マレイン酸部分を構築し、天然物の全合成に必要な全ての炭素骨格および官能基を有する化合物41を得ることができた。  Scheme 10 Scheme 10 現在、CP-263,114の全合成を目指して41の変換を進めており、近い将来、全合成が達成できると確信している。 | 

