学位論文要旨
| No | 126261 | |
| 著者(漢字) | 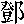 ,穎氷 ,穎氷 | |
| 著者(英字) | Deng,yingbing | |
| 著者(カナ) | トウ,エイヒョウ | |
| 標題(和) | 肝炎ウイルス関連肝癌におけるDNAメチル化の異常 | |
| 標題(洋) | Aberrant DNA methylation of the genes in hepatitis B and C virus-related hepatocellular carcinoma | |
| 報告番号 | 126261 | |
| 報告番号 | 甲26261 | |
| 学位授与日 | 2010.04.21 | |
| 学位種別 | 課程博士 | |
| 学位種類 | 博士(医学) | |
| 学位記番号 | 博医第3552号 | |
| 研究科 | 医学系研究科 | |
| 専攻 | 外科学専攻 | |
| 論文審査委員 | ||
| 内容要旨 | 【Background and Aim】Hepatocellular carcinoma (HCC) is the sixth most common malignancies and the third most common cause of cancer mortality in the world. Among numerous causative factors suggested, chronic infection caused by hepatitis B virus (HBV) and/or hepatitis C virus (HCV) has been defined as the main risk factor for HCC development. Chronic HBV infection is the predominant risk factor for HCC in Southeast Asia and Africa, whereas chronic HCV infection is predominant in Western countries and Japan. HBV, DNA-containing virus, and HCV, RNA-containing virus. They showed different characteristics in carcinogenesis and clinical presentation of HCC. It is reported that about 10% HBV-carriers grow into chronic hepatitis patients, 20% of which transform into liver cirrhosis (LC) and HCC. HBV can also cause HCC in the absence of LC through the pathway of integration into the human genome. On the other hand, more than 60% of HCV-carriers grow into LC and mostly develop to HCC during several decades. The onset of HCV-related HCC is later than that of HBV-related HCC. HCC is caused by genetic or epigenetic alterations. The genetic alterations include chromosomal instability with point mutations and deletions. Chromosomal instability of CDKN2A and TP53 loci was observed in HCC, and mutations of AXIN1 and PIK3CA were more frequently seen in HBV-related HCC than HCV-related HCC. Aberrant DNA methylation of promoter CpG islands has been described as one of the major epigenetic alterations in human cancers, including HCC. It has been reported that methylation of promoter CpG islands of p16 tumor suppressor gene occur in 73% HCC tissues, and in 56% of HBV-related HCC and 84% of HCV-related HCC. Yang et al. reported that methylation of SOCS-1, APC, and p15 was more frequently observed in HCV-related HCC than HBV-related HCC. It may be a consequence that different genetic/epigenetic pathways may be involved in the process of carcinogenesis in HBV-related HCC and HCV-related HCC, and that HBV-related HCC may occur mainly through chromosomal instability, whereas HCV-related HCC may be more involved with aberrant DNA methylation, and there may be genes methylated preferentially to HCV-related HCC. To enable high-resolution mapping of DNA methylation, we reported methylated DNA immunoprecipitation (MeDIP)-chip analysis on short oligonucleotide array using unbiased amplification of MeDIP products by in vitro transcription, and applied the method to genome-wide DNA methylation analysis of colorectal cancer. In the present study, we analyzed aberrant promoter methylation in 6 HCC clinical samples (including 3 HBV-related HCCs and 3 HCV-related HCCs) and noncancerous tissues on genome-wide scale by the method. First aim, candidate regions of promoter methylation preferentially to HBV-related HCC and HCV-related HCC are selected, and the methylation levels of these genes are measured quantitatively using MassARRAY. Second aim, correlation between DNA methylation status and clinicopathological features is analyzed. 【Materials and Methods】Genome-wide DNA methylation status of 6 clinical HCC samples and their matched noncancerous liver tissues and 3 normal livers were analyzed by MeDIP-chip. mRNA expression levels of 10 pairs of HCC were analyzed by expression microarrays. DNA methylation in 125 clinical samples and 5 liver tumor cell lines were analyzed by MassARRAY. If methylation level of a gene was >75% in a cell line, expression level of the gene in the methylated cell line and its reexpression after 5-Aza-2'-deoxycitidine/Trichostatin A treatment were analyzed by real-time RT-PCR. 【Result】Six clinical HCC samples and their matched noncancerous liver tissues and 3 normal livers were analyzed by MeDIP-chip. Genes that possessed regions with P < 10-2 within 1kb ± transcription start site were regarded as candidate genes with promoter methylation. There were 4,444 genes methylated in at least one of six HCCs but neither in any matched noncancerous liver tissues nor in any normal liver tissues. Among these genes, 3,019 genes were methylated only in HCV-related HCC, while 518 genes were methylated only in HBV-related HCC. Aberrantly methylated genes were detected in much higher frequency in HCV-related HCC than HBV-related HCC, suggesting a different methylation status between HBV- and HCV-related HCC. Eighty-nine genes methylated in 3 HCV-related HCCs but no HBV-related HCC were regarded as candidate genes methylated specifically in HCV-related HCC (C-markers, hereafter). Seventy-one genes methylated in 2 of 3 HBV-related HCCs but no HCV-related HCC, and 3 genes methylated in 3 HBV-related HCCs and in only 1 of 3 HCV-related HCC, were regarded as candidate genes methylated specifically in HBV-related HCC (B-markers, hereafter). 18 candidate C-markers and 6 candidate B-markers, were validated for the quantitativity and used for further analysis. Using methylation increase value of HCC compared to the matched noncancerous livers, hierarchical clustering analysis stratified a group of 5 HCV-related HCCs as samples with higher methylation. Expression of these genes was analyzed in 10 pairs of HCC by expression microarrays, and we observed decrease of expression to <0.5-fold mainly occurred in HCV-related HCC samples, suggesting gene silencing by promoter methylaion. To confirm gene silencing by promoter methylation, methylation levels in 5 liver tumor cell lines were analyzed by MassARRAY. If methylation level of a gene was >75% in a cell line, expression level of the gene in the methylated cell line and its reexpression after 5-Aza-2'-deoxycitidine/Trichostatin A treatment were analyzed by real-time RT-PCR. All the analyzed genes showed very low expression level in methylated cell lines, and showed upreglation after 5-Aza-2'-deoxycitidine treatment alone or with Trichostatin A treatment, suggesting that the genes were silenced by promoter methylation in HCC. Neither a cluster of methylated HBV-related HCC nor decreased expression preferentially to HBV-related methylation, was observed. Methylation levels were measured by MassARRAY in 125 clinical samples. All the 18 validated C-markers were significantly more methylated in HCV-related HCC compared with HBV-related HCC. As for the 5 tentative B-markers, however, only 2 genes, CYP7B1 and SMOC2, showed significantly more frequent hypermethylation in HBV-related HCC. Since HCV-related HCC patients were significantly elder, methylation levels were compared between age-matched (51-69 years old) HBV- and HCV-related HCC samples. Among 18 C-markers, 15 markers still showed significant difference of methylation levels, therefore were considered to be preferentially methylated in HCV-related HCCs independent from age. Among the 2 B-markers, only CYP7B1 showed significant difference of methylation in comparison of age-matched HCCs. Hierarchical clustering of 59 HCC samples was performed using the methylation difference values in the 59 HCC samples compared to the matched noncancerous liver samples. When the 15 age-independent C-markers were used, a cluster of frequently methylated HCC was detected, which was significantly correlated to HCV-related HCC. The cluster and its significant correlation to HCV-related HCC indicated that genes methylated preferentially in HCV-related HCC exist, including the 15 identified C-markers at least. DNA methylation status of the 15 C-markers was compared with clinicopathological features including gender, age, background liver tissue, tumor size, tumor differentiation, serum AFP, and serum PIVKAII. Methylation of CYP24A1, DLX1, NPR1, SFRP4, DUSP4 and PARQ8 significantly correlated with older age (>60 years old). As for gender, female correlated with SFRP4 methylation. The other features did not correlated with methylation of any C-markers. About prognosis, correlation between disease-free survival and methylation statuses of the 15 C-markers was analyzed by Kaplan-Meyer method. When whole HCC cases were analyzed, HCCs with DUSP4 methylation and CYP24A1 methylation showed significant correlation with survival without recurrence (P<0.05), and methylation of three other genes, ZNF141, RRAD and SFRP4, showed tendency of better prognosis (P<0.01). But methylation of any genes did not correlate with poorer prognosis. When the correlation was analyzed among HCC-related HCCs only, methylation of DUSP4 and NPR1 significantly correlated with better prognosis (P<0.05). CYP24A1 methylation tended to correlate with better prognosis among HCV-related HCCs (P=0.05). There was no significant difference between prognoses of HBV-related and HCV-related HCCs. 【Discussion】Our results demonstrated that HCV-related HCC and HBV-related HCC could be classified into different groups by DNA methylation information. It was indicated that C-markers exist, at least 15 genes, and it was suggested that DNA methylation might be an important cause of hepatic carcinogenesis resulting from HCV infection. HCV-related HCC patients were significantly older than patients with HBV-related HCC or metastatic liver tumor, so age-dependent methylation may have to be considered. But 15 among 18 C-marker genes were methylated in age-matched HCV-related HCC significantly more than HBV-related HCC. As for HBV, only one gene CYP7B1 was found to be methylated in age-matched HBV-related HCC significantly more than HCV-related HCC, and involvement of promoter methylation was suggested to be smaller in this study. But detection of candidate methylation genes was started using 3 HBV-related and 3 HCV-related HCCs, and further study is necessary to clarify the involvement and importance of DNA methylation in HBV-related HCC, including the absence/existence of B-markers. Genes inhibitory to Ras/Raf/ERK signal (NPR1, DUSP4, LOX and RRAD) and inhibitory to Wnt/β-catenin signal (SFRP4 and RUNX3) were included, suggensting that while methylation of some C-markers may be passenger methylation, methylation of a part of C-markers might contribute to genesis of HCC by disrupting cancer-related pathways through silencing a variety of genes. C-markers also included the detoxification enzyme genes. CYP24A1 and NQO1 were reported in HCC. These two genes belong to phase I/II xenobiotic-metabolizing enzyme family and these enzymes play an important role in protecting cells from cytotoxic and carcinogenic agents. Disruption of the detoxification enzymes might cause excessive reactive oxygen species and result in the initiation of HCC. The reason why genes, at least C-markers, were preferentially methylated in HCV-related HCC is still unknown. HCV core protein was reported to down-regulate expression of E-cadherin correlated with CpG island methylation of E-cadherin promoter through activation of DNMT1 and DNMT3B in HCV core protein-expressing HepG2 cell line. Frequent methylation may occur through activation of DNMT by HCV core protein. Or, one may explain it by longer infection period. HCV patients develop HCC with chronic infection for several decades. HBV can cause HCC at earlier onsets, in the absence of LC through integration into the human genome. Actually HCV-related HCC patients were significantly older than HBV-related HCC patients, so methylation of C-markers was confirmed in age-matched patients. But longer period from infection could give more chances to methylation alteration and still should be considered. Methylation of three C-markers, DUSP4, NPR1 and CYP24A1, correlated with recurrence-free survival. One may suggest two possibilities. First, this may be because C-markers in this study were extracted as genes methylated in tumors only but not in background liver tissues. HCC recurrence could be caused either by intrahepatic metastasis or multicentric occurrence. Specific gene expression pattern in cancerous tissues of HCC could predict early recurrence, whereas expression in the background tissues had also been studied based on the idea of field cancerization and multicentric carcinogenesis. Genes methylated in tumor only is considered to contribute to genesis of the resected tumor and its intrahepatic metastasis, but not to genesis of the multicentric recurrent tumor. The correlation with better prognosis might suggest the importance of milticentric recurrence, and methylation alteration accumulated in the background liver as well as tumor might be a determinant of early multicentric recurrence. Second possibility is, accumulation of DNA methylation may contribute in carcinogenesis but rather cause HCC with better prognosis, while HCCs due to other mechanisms e.g. chromosomal instability may perhaps cause cancer with poorer prognosis, as colorectal cancer with highly frequent methylation correlated with microsatellite instability and therefore with better prognosis. Additional studies are necessary to investigate these possibilities, and to clarify the prognostic significance of C-marker methylation itself using a larger cohort of patients. 【Conclusion】Genome-wide MeDIP-chip analysis of clinical samples and quantitative DNA methylation analysis using MassARRAY were performed, and it was found that genes methylated preferentially in HCV-related HCC exist, at least 15 genes. It was suggested that promoter methylation might play an important role in HCV-related HCC, possibly by inactivating inhibitors for cancer-related pathway, and might perhaps be useful for prognostic marker. | |
| 審査要旨 | 本研究は肝炎ウイルス関連肝癌とプロモーター領域の異常メチル化の関係を理解する上で重要と考えられる。HBV・HCV関連肝癌の異常メチル化状態の違いを明らかにするため、両者におけるプロモーター領域の網羅的メチル化解析を行い、同定したHBV/HCV関連肝癌に特異的なメチル化遺伝子において肝癌発癌のメカニズムとの関係を考察しており、下記の結果を得ている。 1. MeDIP-Chip法を用いて遺伝子のプロモーター領域において、正常肝組織(3例)及びHBV・HCV関連肝癌6例と非癌部6例のDNAのメチル化状態を解析した。解析した結果からHCV関連肝癌の3例でプロモーター領域の異常メチル化が多い傾向を認められた。 2. HBV・HCV関連肝癌10例と非癌部10例を用いて18個のHCV特異的候補遺伝子のメチル化率の値をクラスタリングしたところ、5例HCV関連肝癌は高メチル化グループにクラスターした。発現マイクロアデータにより癌部と非癌部の発現量geneチップスコアを割算したところ、主にHCV関連肝癌で発現低下していることを確認した。HCV関連肝癌でメチル化遺伝子の発現制御の傾向が観察されたが、HBV関連肝癌でメチル化遺伝子の発現制御の傾向を観察されなかった。 3. 肝癌細胞株(Huh7、Huh6、HepG2、Alex、Hep3B)を用いてMassARRAYでメチル化率を測定した。メチル化率75%以上の細胞株で5-Aza/TSAを投与し、その投与前後の遺伝子の発現量をRT-PCR法により比較した。5-Aza/TSAを投与した後に、メチル化していた遺伝子の発現量回復が認められた。B/Cマーカーのサイレンシングの原因はメチル化発現制御機構によるものであることが確認された。 4. 125例の臨床検体を用いてHCV特異的なメチル化候補遺伝子18個とHBV特異的なメチル化候補遺伝子6個のメチル化率をMassARRAYで解析した。HCV特異的なメチル化遺伝子18個はすべてHCV関連肝癌でHBV関連肝癌と比べて有意に高率にメチル化されていた。51歳から69歳までの症例のみを解析に使用し、肝炎ウイルス関連肝癌とメチル化の関連性を比較した。HCV特異的なメチル化遺伝子18個のうち15遺伝子はHCV関連肝癌においてHBV関連肝癌と比べて有意に高率にメチル化されていた。15個の遺伝子は年齢に相関なくHCV関連肝癌特異的にメチル化すると考えられた。59例の肝癌と非癌部のメチル化値の差を利用して、クラスターの解析を行った。年齢に依存しないCマーカー15個が高頻度にメチル化されているクラスターはHCV関連肝癌が有意に多いことを認めた。Bマーカー6個では確認されなかった。少なくとも15個はHCV関連肝癌特異的なメチル化遺伝子と考えられた。 5. Cマーカー15個のうち、NPR1、DUSP4、LOX、RRADの4遺伝子はRas/Raf/ERK シグナルの阻害遺伝子である。また、SFRP4、RUNX3の2遺伝子はWnt/・-cateninシグナルの阻害遺伝子である。HCV関連肝癌においては異常プロモーターメチル化によって発癌シグナル経路Ras、Wntなどの阻害剤が不活性化する可能性が示唆された。 以上、本論文はMeDIP-Chipを用いてゲノムワイドにHBV・HCV関連肝癌のメチル化状態を比較した。HCV関連肝癌では、遺伝子のプロモーター領域における異常なメチル化がHBV関連肝癌と比較し多い傾向が認められた。MassARRAYを使用し多数の臨床サンプルの定量的DNAメチル化解析を行い、MeDIP-Chip結果と同様にプロモーター領域においてHBV関連肝癌よりもHCV関連肝癌でメチル化が異常に亢進していることが示された。プロモーター領域がメチル化され、その結果遺伝子発現が抑制されている、HCV関連肝癌で特異的な遺伝子を同定した。これは、プロモーターメチル化はHCV関連肝癌の発癌の解明に重要な貢献をなすと考えられ、学位の授与に値するものと考えられる。 | |
| UTokyo Repositoryリンク |