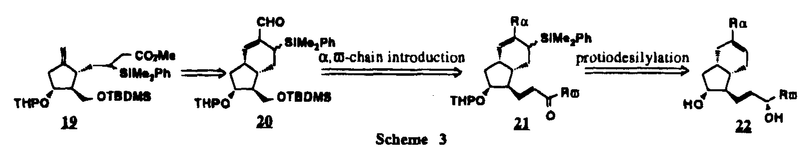
Prostacyclin(PGI2; )は、極めて強力な血小板凝集抑制作用を有するエイコサノイドであり、オータコイドの一つとして生体内の組織・器官に広く分布していることが明らかにされている。は発見当初から循環器領域を中心に医薬品化のターゲットとしてとらえられてきた。しかしながら、は化学的に極めて不安定な化合物であり、また副作用の原因ともなる血管平滑筋の弛緩作用やその他細胞保護作用など種々の作用を有しているため、多様な作用を示しこのまま医薬品として用いるには多くの制限があった。の不安定性の主たる原因は5員環エノールエーテル構造にあるため、この部分を活性を損なうことなく化学的に変換・修飾して安定化を図る合成研究が数多く行われてきた。これらの中で実用化の可能性が期待されるものとして、Carbacyclin( )は、極めて強力な血小板凝集抑制作用を有するエイコサノイドであり、オータコイドの一つとして生体内の組織・器官に広く分布していることが明らかにされている。は発見当初から循環器領域を中心に医薬品化のターゲットとしてとらえられてきた。しかしながら、は化学的に極めて不安定な化合物であり、また副作用の原因ともなる血管平滑筋の弛緩作用やその他細胞保護作用など種々の作用を有しているため、多様な作用を示しこのまま医薬品として用いるには多くの制限があった。の不安定性の主たる原因は5員環エノールエーテル構造にあるため、この部分を活性を損なうことなく化学的に変換・修飾して安定化を図る合成研究が数多く行われてきた。これらの中で実用化の可能性が期待されるものとして、Carbacyclin( )などの炭素アナローグの安定類縁体を挙げることができる。その後、池上・柴崎らによって以上の活性を示すIsocarbacyclin( )などの炭素アナローグの安定類縁体を挙げることができる。その後、池上・柴崎らによって以上の活性を示すIsocarbacyclin(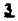 )、続いて柴崎・高橋らによりの母核を環拡大しcis-bicyclo[4.3.0]non-2-eneに変換したHomoisocarbacyclin( )、続いて柴崎・高橋らによりの母核を環拡大しcis-bicyclo[4.3.0]non-2-eneに変換したHomoisocarbacyclin( )が合成された(Fig.1)。の活性はPGI2の1/100程度ではあるが、作用分離の点で優れており注目される。そこで、を化学的に修飾し作用の増強を図るための検討を行った。 )が合成された(Fig.1)。の活性はPGI2の1/100程度ではあるが、作用分離の点で優れており注目される。そこで、を化学的に修飾し作用の増強を図るための検討を行った。 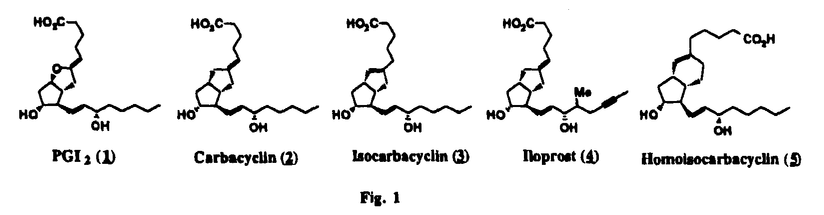 Fig.1 Fig.1 ホモイソカルバサイクリン類縁体の合成 一般に、PG類の および および 側鎖は生体内において種々の酵素によって代謝され速やかに失活することが知られている。そこで、先ず、Homoisocarbacyclin()の及び側鎖の変換・修飾を計画した。安定類縁体の合成は、柴崎らにより開発された方法で、共通鍵中間体としてaldehyde( 側鎖は生体内において種々の酵素によって代謝され速やかに失活することが知られている。そこで、先ず、Homoisocarbacyclin()の及び側鎖の変換・修飾を計画した。安定類縁体の合成は、柴崎らにより開発された方法で、共通鍵中間体としてaldehyde(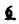 )を用い、これを機軸に種々の及び側鎖を導入する立体選択性に優れた効率的な合成ルートにより進めることとした(Scheme 1,2)。 )を用い、これを機軸に種々の及び側鎖を導入する立体選択性に優れた効率的な合成ルートにより進めることとした(Scheme 1,2)。 (1)側鎖の導入:まず、側鎖の代謝経路である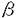 酸化を抑えるため3位を酸素原子に変換した3-オキサ類縁体の合成を行った。鍵中間体のaldehyde()をWittig反応によりジエン( 酸化を抑えるため3位を酸素原子に変換した3-オキサ類縁体の合成を行った。鍵中間体のaldehyde()をWittig反応によりジエン(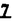 )とし、次いでヒドロホウ素化、酸化してalcoholを得、t-butyl bromoacetateと反応させて3-オキサの側鎖を構築した(Scheme 1)。 )とし、次いでヒドロホウ素化、酸化してalcoholを得、t-butyl bromoacetateと反応させて3-オキサの側鎖を構築した(Scheme 1)。 次に、側鎖に環内二重結合と共役できる二重結合の導入が活性の向上に有効であることが知られていることから、にt-BuOKの存在下3-ethoxycarbonylpropyltriphenyl phosphonium bromideとのWittig反応を行い立体選択的にZ形の共役ジエン側鎖を導入した(Z/E=24/1)(Scheme 1)。 また、活性及び安定性の向上を期待し、3-オキサの側鎖5位に環内二重結合と共役できるエキソメチレン基の導入を試みた。とdimethylisopropoxysilylmethylmagnesium chlorideとのGrignard反応を行い、次いで、H2O2にて処理してdiol(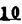 )とし、1級alcoholを選択的に保護した後、2級alcoholの酸化、Wittig反応によるメチレン化、脱保護、さらに、t-butyl bromoacetateと反応させて側鎖を構築した(Scheme 1)。 )とし、1級alcoholを選択的に保護した後、2級alcoholの酸化、Wittig反応によるメチレン化、脱保護、さらに、t-butyl bromoacetateと反応させて側鎖を構築した(Scheme 1)。 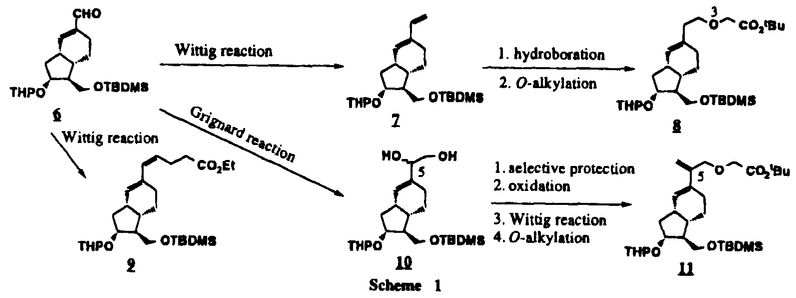 Scheme 1 Scheme 1 (2)側鎖の導入:側鎖導入中間体(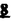 , ,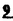 , ,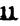 )を脱シリル化、酸化してaldehydeとした後、Emmons-Homer反応により側鎖の構成部分を導入してenone( )を脱シリル化、酸化してaldehydeとした後、Emmons-Homer反応により側鎖の構成部分を導入してenone(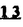 )とし、15位(PG番号)のカルボニル基をNaBH4で還元した後、分離精製して目的の配置alcoholを得、最後にエステル加水分解を行い類縁体( )とし、15位(PG番号)のカルボニル基をNaBH4で還元した後、分離精製して目的の配置alcoholを得、最後にエステル加水分解を行い類縁体(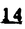 )とした。 )とした。 なお、15位(PG番号)のカルボニル基の還元反応においては、2,6-di-t-butyl-4-methylphenolとDIBAHを用いる山本らの方法により配置のalcoholを選択的に得ることができた(Scheme 2)。 側鎖の修飾の一環として、生理活性を損なうことなく代謝安定性を大幅に向上し得ることが報告されている側鎖13位(PG番号)の三重結合への変換を検討した。側鎖の導入は、柴崎らにより開発された方法で、,-dibromoketoneから発生させた-bromoenolateとaldehydeとのAldol反応を応用し進めた。即ち、側鎖導入中間体(,)をaldehydeに導いた後、Zn-Et2AlCl-CuBrの存在下に,-dibromoketoneとのAldol反応を行い、次いで、MsCl,Et3N,DBUの条件下に脱水反応を行い-bromoenone(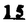 )を得た。はTHP基を脱保護した後、副生したenone( )を得た。はTHP基を脱保護した後、副生したenone(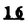 )を分離除去し、これを還元反応に付し15位(PG番号)における-alcoholを分離、さらに、toluene中n-Bu4N・HSO4-50%NaOHにて脱HBr反応を行って側鎖を構築し類縁体( )を分離除去し、これを還元反応に付し15位(PG番号)における-alcoholを分離、さらに、toluene中n-Bu4N・HSO4-50%NaOHにて脱HBr反応を行って側鎖を構築し類縁体(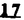 )を合成した。 )を合成した。 また、13位(PG番号)二重結合の単結合への変換を試みた。合成法はenone()を脱保護した後、vitride-CuBrにて還元してketoneとし、次いで上記側鎖の合成ルートと同様に進め目的の類縁体(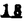 )まで導いた。 )まで導いた。 しかし、これら類縁体()においては15位水酸基のepimerを分離することは困難であった(Scheme 2)。 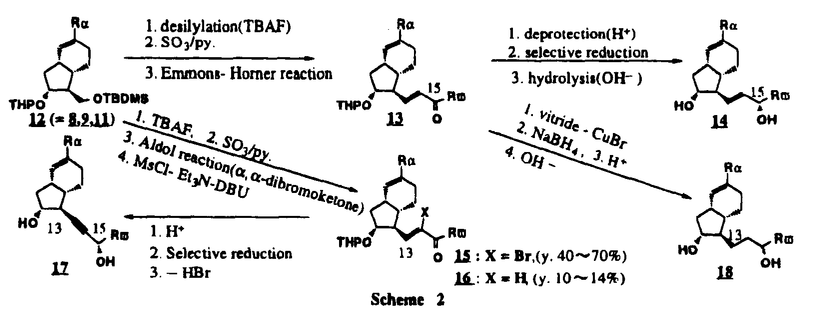 Scheme 2 Scheme 2 (3)シス-ビシクロ[4.3.0]ノナ-3-エン類縁体の合成:Homoisocarbacyclin()の環内オレフィン位置異性体(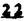 :天然型側鎖)も同様に比較的強い生理活性を示すことが知られていることから、この母核に化学修飾した及び側鎖の導入を試みた。環内二重結合はプロトン化脱シリル化反応をキーステップとして位置選択的に構築したが、3-オキサおよび共役ジエンタイプのいずれの系においても本反応は選択的に進行し、目的の母核を有する類縁体()を好収率で得ることができた(Scheme 3)。 :天然型側鎖)も同様に比較的強い生理活性を示すことが知られていることから、この母核に化学修飾した及び側鎖の導入を試みた。環内二重結合はプロトン化脱シリル化反応をキーステップとして位置選択的に構築したが、3-オキサおよび共役ジエンタイプのいずれの系においても本反応は選択的に進行し、目的の母核を有する類縁体()を好収率で得ることができた(Scheme 3)。 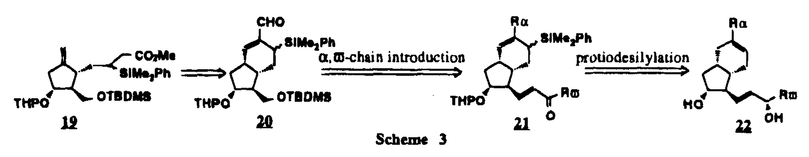 Scheme 3 Scheme 3 側鎖13位三重結合導入の改良合成法の開発 後述のように13位(PG番号)への三重結合の導入はHomoisocarbacyclin類縁体に強力な生理活性を発揮させ、しかも代謝安定性の向上にも寄与する極めて重要な変換であることが明らかとなったが、-bromoenolateを用いるAldol反応においては、-bromoenone()の生成に付随しenone()が副生するという問題があった(Scheme 2)。これは実用的な大量合成を想定した場合、障害になると考えられることから、enone()の副生を抑え得る改良法の開発が必要となった。16はAldol反応において用いるZn-Et2AlCl-CuBrコンプレックスによって、-bromohydrin( )がさらに脱臭素化され副生するものと考えられる。この脱臭素化を回避すべく、より緩和な条件下でのAldol反応を種々検討したが、満足できる結果を得ることはできなかった。そこで、臭素原子よりも還元されにくいと考えられる塩素原子に変え検討することとした(Scheme 4)。 )がさらに脱臭素化され副生するものと考えられる。この脱臭素化を回避すべく、より緩和な条件下でのAldol反応を種々検討したが、満足できる結果を得ることはできなかった。そこで、臭素原子よりも還元されにくいと考えられる塩素原子に変え検討することとした(Scheme 4)。 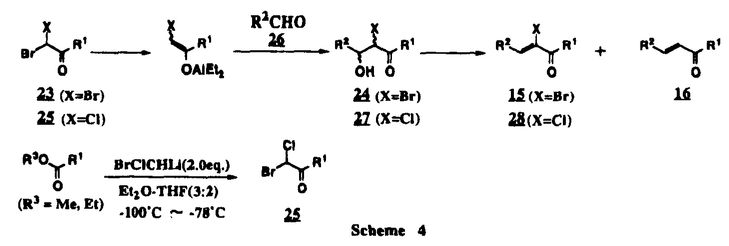 Scheme 4 Scheme 4 esterにBrClCHLiを反応させて得られる-bromo--chloroketone( )とaldehyde( )とaldehyde(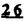 )とのAldol反応を行ない、次いで脱水反応を行ったところ高収率で目的の-chloroenone( )とのAldol反応を行ない、次いで脱水反応を行ったところ高収率で目的の-chloroenone(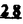 )が得られ、enone()の副生は極めて少量(〜2%)であった。-chloroenone(28j)は脱THP化し立体選択的還元反応の後、分離精製して-alcohol( )が得られ、enone()の副生は極めて少量(〜2%)であった。-chloroenone(28j)は脱THP化し立体選択的還元反応の後、分離精製して-alcohol(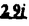 )を得、次いで、脱HCl反応を行ったところ、86%の好収率で目的の13位三重結合導入体()に導くことができた(Scheme 5)。 )を得、次いで、脱HCl反応を行ったところ、86%の好収率で目的の13位三重結合導入体()に導くことができた(Scheme 5)。 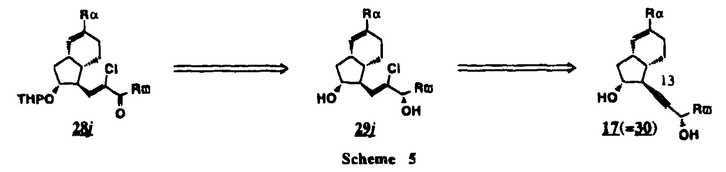 Scheme 5 Scheme 5 本改良法はHomoisocarabacyclinの系ばかりでなく、他のPG系、さらに、それ以外の系でも幅広く応用可能であり、従来法を凌ぐ優れた方法であることが明らかにされると共に効率の良いacetylenic alcohol(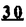 )の一般的合成法になり得ることが確認された(Table I)。 )の一般的合成法になり得ることが確認された(Table I)。 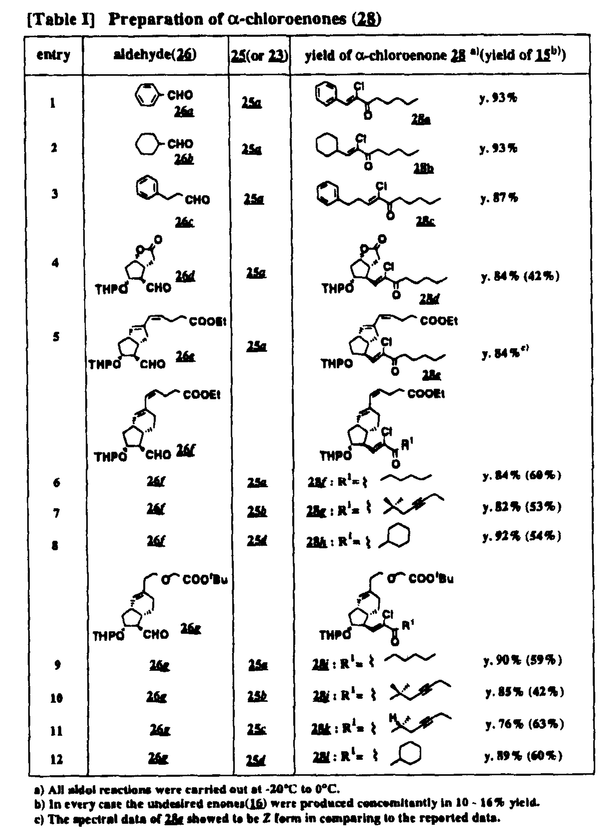 [Table I] Preparation of -chloroenones() [Table I] Preparation of -chloroenones() ところで、本合成法におけるAldol反応、それに続く脱水反応により得られる-chloroenone()はZ体であり、E体の生成は認められなかった。この選択性の理由を明らかにするため、まず、Aldol反応により得られる-chlorohydrinについて立体異性体の生成比[syn体(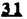 )/anti体( )/anti体(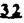 )]を1H-NMRスペクトルで比較検討したが、明確な選択性は認められなかった。次に、単離した-chlorohydrinの立体異性体混合物をMsCl,DBU,Et3Nで処理した結果(Z)-enone( )]を1H-NMRスペクトルで比較検討したが、明確な選択性は認められなかった。次に、単離した-chlorohydrinの立体異性体混合物をMsCl,DBU,Et3Nで処理した結果(Z)-enone(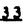 )のみが得られ、Et3Nの存在下に室温で反応すると(E)-epoxyketone( )のみが得られ、Et3Nの存在下に室温で反応すると(E)-epoxyketone(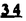 )のみが選択的に得られた。また、分離可能であったsyn体( )のみが選択的に得られた。また、分離可能であったsyn体(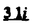 )及びanti体( )及びanti体(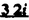 )それぞれを同一条件下に脱水反応を行ったところ、いずれも(Z)-enone( )それぞれを同一条件下に脱水反応を行ったところ、いずれも(Z)-enone(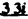 )のみが得られた。これらの実験結果から、Et3Nの存在下においては-chlorohydrinのsyn体()とanti体()は容易に異性化し、安定系の生成物を与えることのできる配置を取り得るため、脱水反応の際しては立体選択的に反応が進行しZ体のみを与えるものと考えられた(Scheme 6)。 )のみが得られた。これらの実験結果から、Et3Nの存在下においては-chlorohydrinのsyn体()とanti体()は容易に異性化し、安定系の生成物を与えることのできる配置を取り得るため、脱水反応の際しては立体選択的に反応が進行しZ体のみを与えるものと考えられた(Scheme 6)。 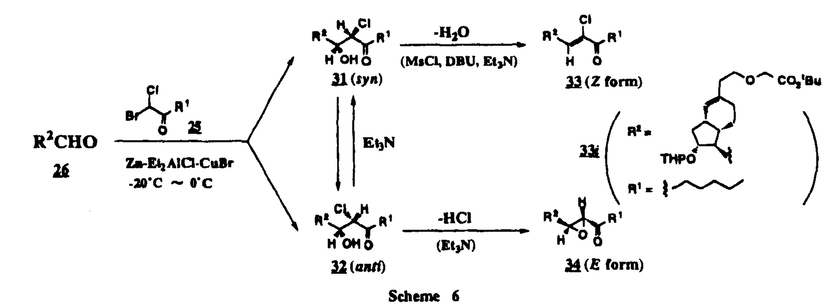 Scheme 6 Scheme 6 ホモイソカルバサイクリン類縁体の生物活性 今回合成したHomoisocarbacyclin類縁体に関する一次スクリーニングとしては、ウサギ洗浄血小板を用い、adenosine diphosphate(ADP)またはcollagenによる血小板凝集誘発に対する抑制作用、及びラットでのEtOH経口投与による胃粘膜損傷に対する保護効果を試験し評価した。 その結果、3-オキサ類縁体( )及び役ジエン側鎖を有する類縁体( )及び役ジエン側鎖を有する類縁体( )共に活性の向上(数倍〜400倍)が見られた。3-オキサ類縁体のうち側鎖にphenoxymethyl基を導入した類縁体( )共に活性の向上(数倍〜400倍)が見られた。3-オキサ類縁体のうち側鎖にphenoxymethyl基を導入した類縁体(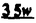 )では強い胃粘膜保護作用を示すが、血小板凝集抑制作用はほとんど示さず、作用の分離の点で特徴が認められた。また、側鎖5位にエキソメチレン基を導入した類縁体においても活性の向上が認められた。 )では強い胃粘膜保護作用を示すが、血小板凝集抑制作用はほとんど示さず、作用の分離の点で特徴が認められた。また、側鎖5位にエキソメチレン基を導入した類縁体においても活性の向上が認められた。 側鎖修飾の検討においては13位デヒドロ体(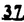 , ,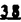 )及び13,14-ジヒドロ体( )及び13,14-ジヒドロ体(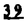 )共に多くの類縁体で生理活性の改善が見られ、極めて強力な血小板凝集抑制作用(IC50:10-10Mレベル)や強力なcytoprotectionを示す類縁体(ED50=1 )共に多くの類縁体で生理活性の改善が見られ、極めて強力な血小板凝集抑制作用(IC50:10-10Mレベル)や強力なcytoprotectionを示す類縁体(ED50=1 g/kg以下)も見出された。さらに、環内二重結合の位置異性体であるcis-bicyclo[4.3.0]non-3-eneを母核とする類縁体も強い活性を示し、この系においては活性が保持されることが明らかになった。 g/kg以下)も見出された。さらに、環内二重結合の位置異性体であるcis-bicyclo[4.3.0]non-3-eneを母核とする類縁体も強い活性を示し、この系においては活性が保持されることが明らかになった。 続いて、強力な活性を示した数個の類縁体については、ウサギを用いex vivoでの経時的血小板凝集抑制作用及び血圧降下作用を試験し、さらに、下痢誘発作用や安定性についても検討を行い総合的に評価した(Table II)。その結果、類縁体(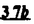 )が最も優れており、開発候補品として選択された。は経口投与の試験においても6時間以上に亘る持続的な作用が確認され、経口投与剤としての可能性も示唆された。 )が最も優れており、開発候補品として選択された。は経口投与の試験においても6時間以上に亘る持続的な作用が確認され、経口投与剤としての可能性も示唆された。 一方、EtOH胃粘膜損傷において、強力な作用が認められた類縁体及び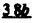 については、数種の実験潰瘍モデルを用い抗潰瘍作用及び酸分泌抑制作用を評価した。両類縁体とも既存のPG系抗潰瘍剤と比較し遜色のない強力な作用を示し、抗潰瘍剤としての可能性が示唆された。 については、数種の実験潰瘍モデルを用い抗潰瘍作用及び酸分泌抑制作用を評価した。両類縁体とも既存のPG系抗潰瘍剤と比較し遜色のない強力な作用を示し、抗潰瘍剤としての可能性が示唆された。 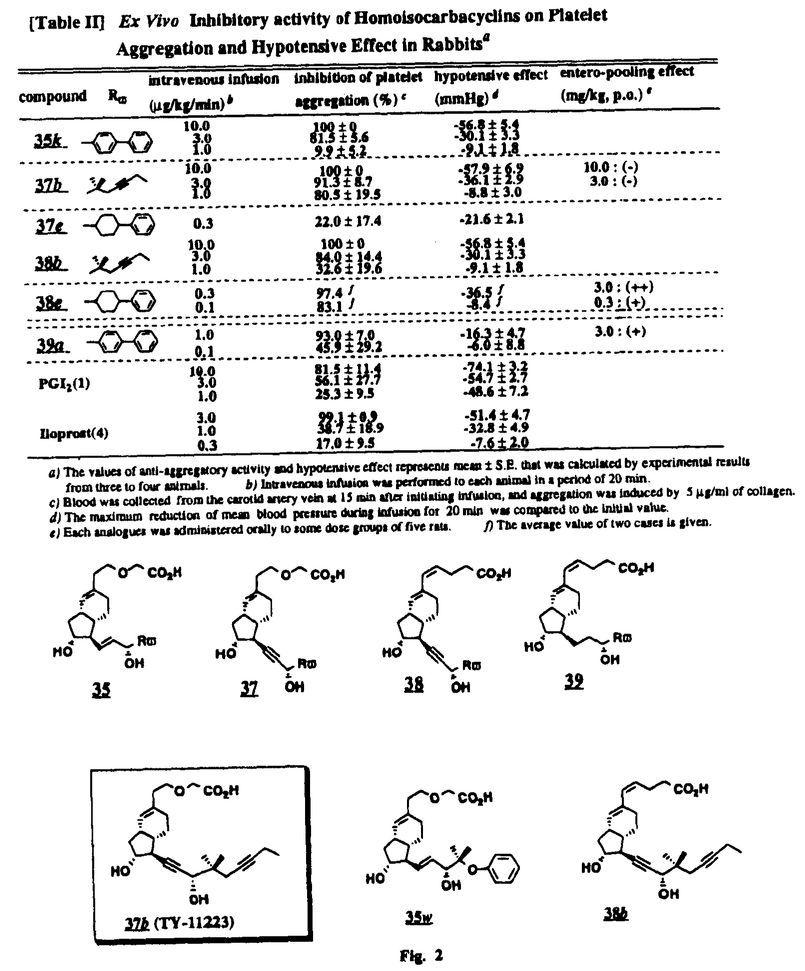 図表[Table II] Ex Vivo Inhibitory activity of Homoisocarbacyclins on Platelet Aggregation and Hypotensive Effect in Rabbitsa / Fig.2 図表[Table II] Ex Vivo Inhibitory activity of Homoisocarbacyclins on Platelet Aggregation and Hypotensive Effect in Rabbitsa / Fig.2 以上、物理化学的な安定性、代謝安定性さらには作用の選択性の向上をめざし、cis-bicyclo[4.3.0]noneneを母核とするHomoisocarbacyclin類縁体の合成研究を行った。その中から従来にない優れた性質を有する類縁体(:TY-11223)を見出すことができ、また、大量合成が可能な効率的改良法を開発し完成させることができた。なお、本化合物については、現在、開発に向け種々の試験が進められているところである。 | 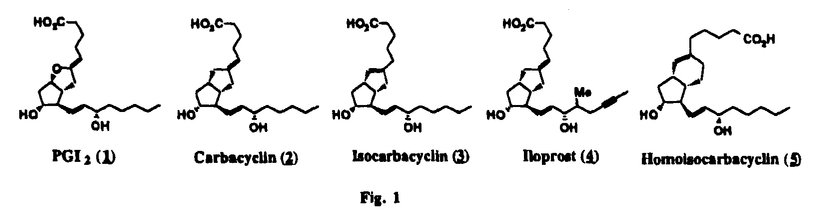
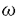 側鎖は生体内において種々の酵素によって代謝され速やかに失活することが知られている。そこで、先ず、
側鎖は生体内において種々の酵素によって代謝され速やかに失活することが知られている。そこで、先ず、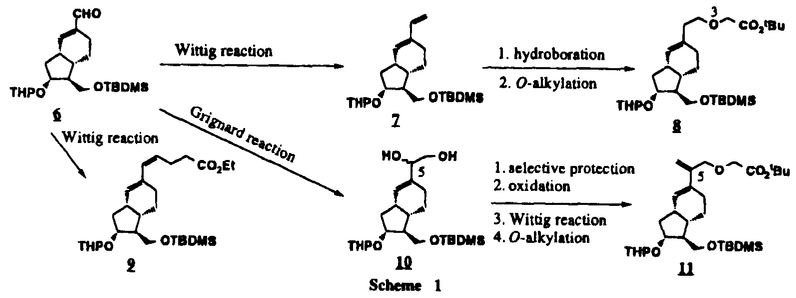
 )がさらに脱臭素化され副生するものと考えられる。この脱臭素化を回避すべく、より緩和な条件下でのAldol反応を種々検討したが、満足できる結果を得ることはできなかった。そこで、臭素原子よりも還元されにくいと考えられる塩素原子に変え検討し、本問題点を解決した(Scheme 3)。
)がさらに脱臭素化され副生するものと考えられる。この脱臭素化を回避すべく、より緩和な条件下でのAldol反応を種々検討したが、満足できる結果を得ることはできなかった。そこで、臭素原子よりも還元されにくいと考えられる塩素原子に変え検討し、本問題点を解決した(Scheme 3)。