天然に広汎に存在するインドールアルカロイドやインドール骨格をもつ医薬、農薬の中には、そのベンゼン環部に置換基を有するものが多く存在し、これらは一般に顕著でかつ多彩な薬理作用を示す。インドール核上ベンゼン部位における官能基の種類と置換位置はその物質の活性発現に大きな影響を与えることから、生理活性インドール類の創製研究を進めるうえで、ベンゼン部位での高い置換位置選択性を有するインドール合成反応の開発は非常に重要な課題である。さて、ベンゼン部置換インドール類の合成に関しては、従来より置換ベンゼン誘導体からFischer法、Reissert法、Leimgruber-Batcho法等によりピロール環部を閉環する方法が用いられていた。しかしながら、これらの多くはベンゼン誘導体の調製において置換位置の任意性および官能基化に限界があり、またインドール閉環反応での位置選択性も問題となる。さらに、苛酷な反応条件のため持てる置換基が限られる場合がある。 これに対し、乙卯研究所の夏目らはピロールを出発原料としたベンゼン部位アルキル置換インドール類の調製法を報告してきた(Chart 1)。これはピロールから導かれる前駆体1,2a,b,3に対し緩和な酸処理でベンゼン部分の閉環を行うことにより、ベンゼン環上位置選択的にアルキル化されたインドール類4を高収率で合成できるという、有用性の高い方法である。著者はこれを更に展開し、ベンゼン部置換インドール類の効率的合成を目指して研究を行い、以下に述べる[I]-[III]の結果を得た。  Chart 1[I]4位モノアルキル置換インドール類の効率的合成 Chart 1[I]4位モノアルキル置換インドール類の効率的合成 夏目らの従来法では、閉環前駆体8の置換基Rは5に対するFriedel-Crafts反応で得られるアシル体6に由来しているため(5→6→8)、その官能基化に大きな制約があった(Chart 2)。これを改善すべく、著者は8の合成のための共通中間体として7を設定し、7に対し置換基Rを種々官能基化の容易なcarbanionとして導入する事とした。  Chart 2 Chart 2 Table1に示すように、7a,bに対し各種官能基化された炭素求核剤(R-)を作用することにより多彩な側鎖を有する閉環前駆体8a,bを高収率で調製できた。次いで、8a,bをその側鎖に応じた酸条件で処理して、対応する4位置換インドール誘導体9a,bを収率良く合成することができた。ここで得た10(run 3)、11(run 4)、12(run 7)はいずれもインドールアルカロイドの重要合成中間体として利用されている化合物である。また、従来11工程を要して合成されていたドーパミン・アゴニスト作用を有する4-[2-(dipropylamino)ethyl)indole[DPAI(14)]は本法を利用すると、13(run8)のLiAlH4処理により1-(phenylsulfonyl)pyrrole(5a)から僅か5工程、51%の高通算収率で得ることができた(Chart3)。以上のように、7を共通中間体とする本反応により、多種多様な4位モノアルキル置換インドール類の簡便な合成が可能となった。 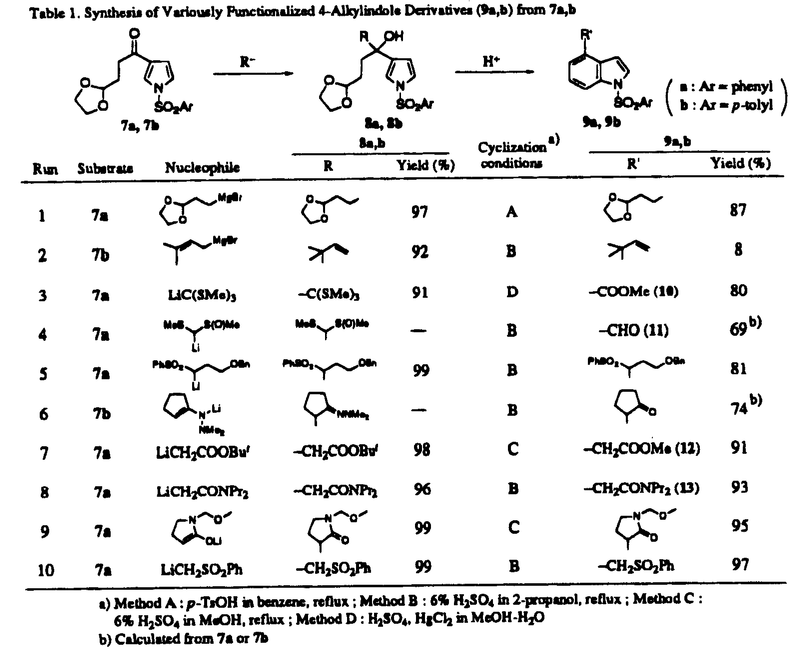 Table 1.Synthesis of Variously Functionalized 4-Alkylindole Derivatives(9a,b)from 7a,b Table 1.Synthesis of Variously Functionalized 4-Alkylindole Derivatives(9a,b)from 7a,b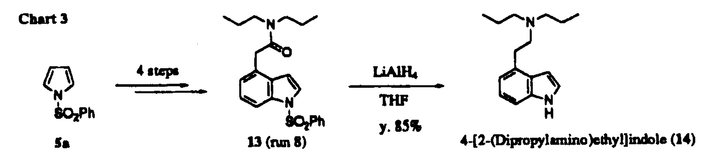 Chart 3[II]ベンゼン部位モノアルコキシ置換インドール類の効率的合成 Chart 3[II]ベンゼン部位モノアルコキシ置換インドール類の効率的合成 [I]で用いた共通中間体7に求核剤を導入することなく直接閉環出来れば、4-ヒドロキシインドール誘導体の合成が可能になる。そこで、4位モノアルコキシインドール類の合成法の確立を目的としてこの閉環反応を種々検討し、Table2に示すような結果を得た。7bを触媒量のp-TsOH存在下ベンゼン中で加熱還流すると、4-アルコキシインドール誘導体17bが45%の収率で生成した(run1)。17bにおける4位置換基は7bのエチレンアセタールに由来する故、次に基質7aについて10等量のエチレングリコールを添加して同条件下反応を行ったところ、17aの収率は90%と飛躍的に向上した(run2)。4-ヒドロキシ体を合成すべく、7のアセタールをアルデヒドとした基質を用いてアルコール無添加で酸処理を行ったが、目的物は全く得られず、複雑な混合物を生じるだけであった。以上の事から、本反応の進行にはアルコールの共存が必須であることが判明した。そこで著者は基質として7a,b,15を用いてこの反応を行い、種々の4-アルコキシインドール誘導体の合成を行った(run3〜8)。過剰のアルコール共存下ベンゼン或いはトルエン中触媒量のp-TsOHで、またはジクロロメタン或いは1,2-dichloroethane中触媒量の硫酸で加熱還流することにより、いずれの場合も用いたアルコールが4位に置換したインドール誘導体16を良好な収率で得ることができた。  Table 2.a) Formation of 4-Alkoxyindole Derivatives(16)from 7a,7b,and 15 Table 2.a) Formation of 4-Alkoxyindole Derivatives(16)from 7a,7b,and 15 次に、この反応を5-,6-,および7-モノアルコキシインドール類の合成へ展開した。この場合、5,6,7位に相当する炭素がカルボニル基化された化合物18,19,20をそれぞれ閉環前駆体として設定し、これらを用いて酸触媒モノアルコキシインドール類の合成反応を検討した(Table3)。18,19に関しては、前述の4-アルコキシ体合成の場合と異なり、アルコールを共存せず10%HCl-DME(1:2)中、50℃で加熱することにより直接それぞれ5-および6-ヒドロキシ体へ変換できた(run1,3)。また、6-メトキシ置換体を合成すべく基質19に4位置換体の調製の際用いた条件(H+/MeOH)を適用したところ、目的物の他に6-ヒドロキシ置換体が相当量副生した(run5)。しかしながら、これは上記の条件に過剰量のCH(OMe)3を添加することにより改善でき、各位メトキシ置換体のみを高収率で得ることができた(run2,4,7)。  Table 3.Formation of 5-,6-,and 7-Alkoxyindole Derivatives(21)from 18,19,and 20[III-1](±)-及び(S)-(-)-Pindololの効率的合成 Table 3.Formation of 5-,6-,and 7-Alkoxyindole Derivatives(21)from 18,19,and 20[III-1](±)-及び(S)-(-)-Pindololの効率的合成 Pindololは高血圧症、不整脈の治療薬として現在、臨床使用されている -遮断薬で、構造上の特徴としてインドール核4位に不斉炭素一つを含むアルコキシル基を有している。Pindolol自体はラセミ化合物であるが、これまでの研究から(S)-(-)-体の方が薬理学的により活性が強いことが知られている。さて、従来のpindolol(22)の合成法は空気酸化等に鋭敏な4-hydroxyindole(24)を使用するという欠点があった(Chart4)。また、(S)-体(23)の合成の際には24と(-)-epichlorohydrin(25)との反応が2つの経路(path aとb)を採りうるため、最終的に(S)-(-)-pindololの光学純度が低下すると報告されている。 -遮断薬で、構造上の特徴としてインドール核4位に不斉炭素一つを含むアルコキシル基を有している。Pindolol自体はラセミ化合物であるが、これまでの研究から(S)-(-)-体の方が薬理学的により活性が強いことが知られている。さて、従来のpindolol(22)の合成法は空気酸化等に鋭敏な4-hydroxyindole(24)を使用するという欠点があった(Chart4)。また、(S)-体(23)の合成の際には24と(-)-epichlorohydrin(25)との反応が2つの経路(path aとb)を採りうるため、最終的に(S)-(-)-pindololの光学純度が低下すると報告されている。  Chart 4 Chart 4 今回著者は[II]で得た知見を応用して、上記の問題点をすべて解決することができた。(±)-pindololの合成を意図して、添加アルコールとして3-chloro-1,2-propanediol(27)を選び、前述のインドール閉環反応によりpindololへ誘導可能な28が効率良く生成する条件を検討した(Chart5)。その結果、26と8当量の27の混合物を硫酸触媒でジクロロメタン中、4時間加熱還流した時、28を位置異性体29に優先して最高収率で得ることができた。28はLiI存在下isopropylamineで処理し30を経由した後、加水分解するか、あるいは一段階で28からisopropylamine共存下、NaOHの作用により(±)-pindolol(22)へ変換できた。  Chart 5 Chart 5 次に、(S)-(-)-pindololの合成を行った。光学活性アルコールとしては、入手が容易で、脂溶性が高く、望みの絶対配置を有する(R)-1-O-toluenesulfonylglycerol(31)を選び、32への変換を検討した(Chart6)。この場合も(±)-体の合成と同様、26を4当量の31と触媒量の硫酸存在下、ジクロロメタン中で3時間加熱還流した時、若干の位置異性体を生じるものの最高収率で32を得ることができた。次いで、32をアミノリシス、加水分解反応に付すことにより、高い光学純度をもつ(S)-(-)-pindolol(23)を合成できた。以上のように、化学的に不安定な4-hydroxyindole(24)を経由せず、また(S)-体の合成では反応途中のラセミ化を一切生じないという特長を有する本合成法により、(±)-及び(S)-(-)-pindololの効率的合成が可能となった。  Chart 6[III-2]4-[2-(Dipropylamino)ethyl]-6-hydroxyindoleの効率的合成 Chart 6[III-2]4-[2-(Dipropylamino)ethyl]-6-hydroxyindoleの効率的合成 4-[2-(dipropylamino)ethyl]-6-hydroxyindole[以下、6-hydroxy-DPAI(41)]は[I]で合成したドーパミン・アゴニスト、DPAI(14)の推定生体内代謝物で、その活性は14よりも強力であるとされている。41を合成するには、4位アルキル側鎖と6位水酸基を位置選択的に導入する必要がある。これまでに二つの合成報告例があるが、いずれも置換ベンゼン誘導体からのインドール合成を利用するもので、多工程を要していた。著者は41をこれまでの知見を基に効率的に合成した(Chart7)。まず、34から3工程(methyl 3-mitropropionateとの縮合、Nef変法、ケタール化)で37を得た。この37とN,N-dipropylacetamide lithium enolateとの縮合反応を過剰のt-BuOK存在下で行うことにより、好収率で閉環前駆体38を得た。次に、前述の6-hydroxyindole誘導体の合成法に倣い、38を過剰量の1,3-propanediol存在下、p-TsOHとトルエン中加熱還流することにより、インドール閉環体39を収率良く得ることができた。最後に側鎖を脱保護し40とした後、LiAlH4でアミドの還元とベンゼンスルホニル基の除去を同時に行い、目的の6-hydroxy-DPAI(41)を34から7工程、通算収率11.5%で合成することができた。  Chart 7 Chart 7 以上、著者は高い電子過剰系のピロールの窒素原子をトシル基、ベンゼンスルホニル基で保護し、ピロール環に適度なnucleophileとしての性質と酸に対する安定性を併わせ持たせる事により、ピロール誘導体からの4位モノアルキル、およびベンゼン部位モノアルコキシ置換インドール形成反応を確立できた。また本法を利用し、(±)-および(S)-(-)-pindolol,更に6-hydroxy-DPAIの効率的合成に成功した。 | 
