パラメーターを含まない第一原理による電子構造計算は、密度汎関数理論が提出されて以来大きな成功をおさめてきた。最近では原子の種類を与えれば、多くの系でその基底状態の結晶系、格子定数、凝集エネルギー、弾性定数などを理論的に決定することができる。 しかし半導体・絶縁体のハンド・ギャップを理論的に正しく決定することが出来ず、実験値の50〜70%程度の値しか与えることができないことは広く知られている。また反強磁性遷移金属酸化物、銅酸化物などいわゆる強相間電子系の基底状態を正しく与えることも出来ない。電子構造を正しく知り種々の物性パラメーターを決定することが多くの議論の出発点であることを考えると、これらの事は重大な問題である。 著者は本論文において、反強磁性絶縁体の基底状態を正しく記述する1電子状態を得るために、局所密度近似に対する自己相互作用補正を、線形マフィンティン軌道法のもとで定式化した。さらにこの定式化を固体水素、遷移金属酸化物に適用し、また一般的に自己相互作用補正を行うための指導原理を提案した。本論文は1〜7章および全体のまとめの計8章からなる。 第1章は磁性のバンド理論およびスレーターとモットの議論について述べさらにスピン密度汎関数法の問題点を指摘し、全体の序となっている。 第2章はスピン密度汎関数法およびその局所密度近似の解説であり、さらに1電子固有エネルギーの意味を、クープマンスの定理(ヤナックの定理)および一般的な自己エネルギーなどの種々の点から明確にしている。 第3章は線形マフィンティン軌道法と、その局在軌道の定式化について述べ、本論文で局在軌道を用いることの意味を論じている。 第4章は密度汎関数理論の局所密度近似の問題点とその対応策としての自己相互作用補正を説明し、原子系における問題点を述べたあと、マフィンティン軌道法における定式化にあてられる。 第5章は固体水素の磁性を、以上の定式化のもとで理論的にとり扱っている。その結果、自己相互作用補正を行ったエネルギーが、使用する交換相関エネルギーの局所密度汎関数の形に強く依存すること、またそのために金属-絶縁体転移を論ずるのは適当でないことが述べられている。特に自己相互作用補正が引力ポテンシャルを与える時でも全エネルギーは上昇する可能性があることを示した点は重大である。一方、反強磁性状態の側では磁気モーメント、バンドギャップなど著しく改善されることが示された。 第6章、この章が本論文の中心をなす。遷移金属酸化物に自己相互作用補正を行った計算結果を示し詳しく議論している。自己相互作用補正を行わないもの(タイプ1)、すべての軌道に自己相互作用補正を行ったもの(タイプ2)、遷移金属元素の軌道にのみ自己相互作用補正を行ったもの(タイプ3)の3つについて計算を行った。それぞれについて、磁気モーメント、バンドギャップ、凝集エネルギーおよび状態密度とバンドを示している。Svaneらは凝集エネルギーが最低になるという条件からタイプ3のものを求め議論している。本論文では、自己相互作用補正を行ったコーン・シャム方程式は変分原理に立っているが、エネルギー値そのものは3つのタイプのどれが安定という精度を持たないことを詳細に論じ、絶縁体には常にタイプ2で計算を行うべきであると結論している。その結果、磁気モーメントおよび占有状態のスペクトルにおける特徴は実験を良く説明するが、バンドギャップについての改善は万全ではない。またVOについては、実験的には常磁性金属となり、今の自己相互作用補正のわく内では議論できない。 第7章は、交換相関エネルギーの関数形や、自己相互作用ポテンシャルの非球対称成分の寄与について詳しく解析している。これにより自己相互作用補正を正しく行うためには局所密度汎関数の形が本質点であるとともに、この理論の枠内では金属・絶縁体転移と磁気転移の間に、特別の関係がないことが明らかに示されている。 以上本論文は、密度汎関数理論における局所密度近似に対する自己相互作用補正の定式化を行い、現実の反強磁性絶縁体の電子状態を論じ、理論の枠組をより一般的に拡張するとともにその限界も明確にした。これは今後の固体物理学の基礎分野に貢献すること大である。よって本論文は博士(工学)の学位請求論文として合格と認める。 | 
 に対して変分すると、次式のような有効ポテンシャル中の1電子方程式が得られる。
に対して変分すると、次式のような有効ポテンシャル中の1電子方程式が得られる。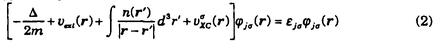
 (r)はLSD法における交換相関ポテンシャルである。LSD法の問題点の1つは、ハートリー・フォック法では静電エネルギーと交換エネルギーで厳密に打ち消し合っていた自己相互作用が、交換相関エネルギーを局所近似したために有限に残ってしまうことである。このLSD法の欠陥を改良する方法がSICであり、(1)式の代わりに次式のエネルギー汎関数を用いる。
(r)はLSD法における交換相関ポテンシャルである。LSD法の問題点の1つは、ハートリー・フォック法では静電エネルギーと交換エネルギーで厳密に打ち消し合っていた自己相互作用が、交換相関エネルギーを局所近似したために有限に残ってしまうことである。このLSD法の欠陥を改良する方法がSICであり、(1)式の代わりに次式のエネルギー汎関数を用いる。
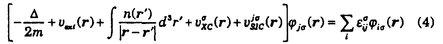
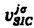 (r)は、軌道j
(r)は、軌道j に対する自己相互作用を補正するSICポテンシャルである。
に対する自己相互作用を補正するSICポテンシャルである。 の非対角項を無視することができない。1電子励起エネルギーは
の非対角項を無視することができない。1電子励起エネルギーは