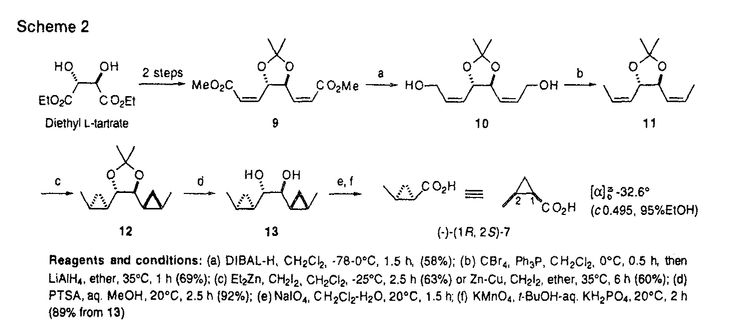
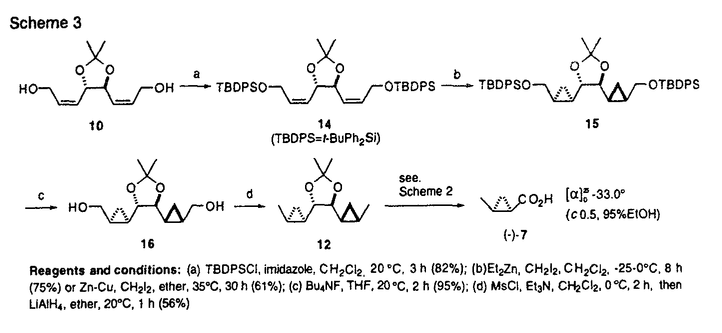
本論文は有糸分裂阻害剤Curacin Aの全合成を主軸として展開した合成研究を記述したものである。 有糸分裂阻害剤は近年、癌の分子標的としても注目されてきたチュブリン蛋白に結合してその重合・解重合を阻害することによって有糸分裂を阻害する化合物群である。有糸分裂阻害剤のチュブリンへの結合部位は、コルヒチン部位とビンブラスチン部位の2つがあり、特に前者を作用点とする有糸分裂阻害剤はそのほとんどが互いに共通の構造的特徴を有する。こうした状況下で近年見いだされた天然物であるCuracin Aは、強力な活性を持ちながら、その構造が従来のコルヒチン部位結合性有糸分裂阻害剤とは全く異なるユニークな化合物として注目されている。よって、Curacin Aならびにその類縁化合物の合成・合成法の確立は、チュブリンを中心に据えた有糸分裂阻害剤の研究領域に大きな寄与を果たす。 本論文に記された研究により、(1)当時未決定であったCuracin Aのチアゾリン骨格部分の絶対構造の推定、(2)効率的なcis-2-メチルシクロプロパン環の不斉合成法の確立、(3)Curacin Aの不斉全合成の達成、(4)p-メトキシベンジルの選択的脱保護法の開発、が成された。加えて本論文には、得られた様々な誘導体のチュブリン重合阻害活性を評価することによる、活性発現のためのにCuracin Aの炭化水素鎖-複素環両部分の必要性、という、従来の予想を覆す発見が記述されている。 第1項に関しては、チアゾリン環部分の考えられるすべての立体異性体をすべて合成し、それらの光学的データを天然Curacin Aのそれと比較することにより達成した。正しくCuracin Aの絶対構造を推定することに成功した。 第2項に関しては、L-酒石酸ジエチルを出発原料とし、新たに開発した不斉ダブルSimmons-Smithシクロプロパン化によって、一分子の簡単な原料からその炭素すべてを利用して2分子の目的物を得るという、効率的な合成法を確立した。 第3項に関しては、L-システイン由来以外の3つの不斉点を完全に立体制御して全合成を達成している。現時点では、本論文以外に5グループからCuracin Aの全合成が報告されているが、その中でも本論文記載の方法は特長ある優れたものと評価できる。 第4項に関しては、ハード酸・ソフト求核剤組み合わせ系のMgBr2OEt2-Me2S反応剤をデザイン・開発している。本方法はいまだ改良の余地ありとはいえ、選択的かつ一般性ある方法であると評価できる。 論文記載の考察の部では、チュブリンを標的とする有糸分裂阻害剤に関する基礎研究・応用研究、さらには、その医薬開発研究に関しての方向性・提案が述べられている。 以上、本論文内容は、研究素材の設定、問題の設定とその解決、得られた結果、いずれにおいても優秀であり、合成化学、天然物化学の発展に寄与するところ大であり博士(薬学)の学位に値するものと判定した。 | 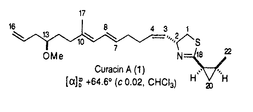



 Mであったが、チアゾリン環を有する部分構造2のいずれの異性体にも阻害活性が見られなかった。さらにホスホニウム塩26とアセトアルデヒドのWittig反応により得られたテトラエン28、及び幾つかの側鎖由来の部分構造に阻害活性が見られなかったことから、その活性発現にはヘテロ環部分と脂溶性側鎖の組み合わせが重要であることをデータにより初めて明らかにした。
Mであったが、チアゾリン環を有する部分構造2のいずれの異性体にも阻害活性が見られなかった。さらにホスホニウム塩26とアセトアルデヒドのWittig反応により得られたテトラエン28、及び幾つかの側鎖由来の部分構造に阻害活性が見られなかったことから、その活性発現にはヘテロ環部分と脂溶性側鎖の組み合わせが重要であることをデータにより初めて明らかにした。