インスリンには、筋肉や脂肪細胞といった末梢組織において、主にGLUT4糖輸送担体を細胞表面に移動(translocation)させ、糖取り込みを増加させる作用があることが知られているが、その作用の詳細な分子メカニズムは明らかでない。その一因として、脂肪細胞などの分化した細胞では、従来、外から遺伝子を導入し発現させることが困難であったことがあげられる。そこで今回、アデノウィルスベクターによる遺伝子導入系を用い、3T3-L1脂肪細胞に以下の3種の蛋白をそれぞれ過剰発現させ、それらの糖輸送機能や糖輸送担体のtranslocationに及ぼす影響を観察することから、インスリンによる糖取り込み促進作用の機構の解明を試みた。 1.細胞内でのphosphatidylinositol(PI)3キナーゼの活性化が重要な役割を果たしていることは、その特異的阻害剤を用いた実験から示唆されている。そこで、PI3キナーゼの調節サブユニットp85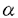 の触媒サブユニットと結合できなくした変異体( の触媒サブユニットと結合できなくした変異体( p85)を過剰発現させ、インスリン受容体から二量体PI3キナーゼへのシグナルを遮断することにより、PI3キナーゼ活性と糖輸送活性との関連を詳細に検討した。 p85)を過剰発現させ、インスリン受容体から二量体PI3キナーゼへのシグナルを遮断することにより、PI3キナーゼ活性と糖輸送活性との関連を詳細に検討した。 2.インスリンは低分子G蛋白Rasを介してMAPキナーゼカスケードをも活性化することが知られている。また、近年、受容体チロシンキナーゼからRasを介してPI3キナーゼが活性化されうることが報告された。そこで、これらの系が、インスリン刺激による糖輸送促進にどの程度関与しているのか確かめる目的で、dominant negative Ras(N17Ras)の過剰発現を試みた。 3.PI3キナーゼ活性化がインスリンの糖輸送促進作用に必須であると考えられているが、それのみで糖輸送促進が惹き起こされるかどうかは明らかではない。そこで、PI3キナーゼの触媒サブユニットであるp110を過剰発現させることによりインスリン受容体を介さずにPI3キナーゼの活性を増加させ、糖輸送活性化が惹き起こされるか検討した。 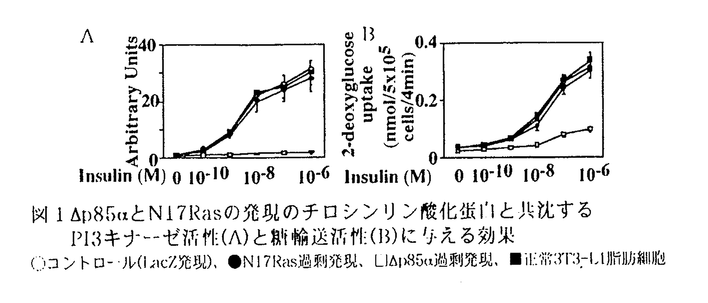 図1p85とN17Rasの発現のチロシンリン酸化蛋白と共沈するPI3キナーゼ活性(A)と糖輸送活性(B)に与える効果○コントロール(LacZ発現)、●N17Ras過剰発現、□p85過剰発現、■正常3T3-L1脂肪細胞 図1p85とN17Rasの発現のチロシンリン酸化蛋白と共沈するPI3キナーゼ活性(A)と糖輸送活性(B)に与える効果○コントロール(LacZ発現)、●N17Ras過剰発現、□p85過剰発現、■正常3T3-L1脂肪細胞 結果 (1)p85PI3キナーゼとRasのdominant negative変異体の発現 1.これら変異蛋白の過剰発現は、それぞれp85PI3キナーゼ、並びに、Ras対する抗体によるimmunobottingにより確認された。 2.p85を過剰発現させた細胞では、インスリン刺激によるチロシンリン酸化蛋白と共沈するPI3キナーゼ活性は著明に抑制された(コントロールの約6%)(図1A)が、MAPキナーゼ活性はコントロールの細胞と同様、インスリン刺激により活性化された(約8倍)。また、N17Rasの過剰発現させると、インスリン刺激によるMAPキナーゼ活性は著明に抑制された(約1.2倍)が、PI3キナーゼ活性には有意な影響は及ぼさなかった。以上より、これらdominant negative変異体は細胞内で機能的に発現していることが確認された。 3.2-デオキシグルコース取り込み能は、N17Ras過剰発現細胞では、コントロールと同様、インスリン刺激により約10倍の活性増加を認めたが、p85過剰発現細胞では、コントロールの約27%にまで抑制された。(図1B)しかし、その抑制の制度はチロシンリン酸化蛋白と共沈するPI3キナーゼ活性の抑制に比べれば弱く、また、p85を過剰発現させた細胞を高濃度(10-7M,106M)のインスリンで刺激した場合、コントロール細胞を低濃度(1010M,109M)のインスリンで刺激した時に比べ、PI3キナーゼ活性は低いにもかかわらず2-デオキシグルコース取り込み活性は高く、PI3キナーゼ活性と糖取り込み活性との間に乖離が認められた。(図1) 4.細胞全体をサンプルとして、SDS-PAGEに流し、GLUT4の抗体でimmunoblotしたところ、GLUT4糖輸送担体の発現はコントロール、p85過剰発現、N17Ras過剰発現3T3-L1脂肪細胞の間では、明確な差は認めなかった。遠心分離によるsubcellular fractionation法により、インスリン刺激によるGLUT4の細胞内移動を検討したところ、N17Rasの発現では、インスリン刺激によるGLUT4のplasma membrane分画への移動はコントロール同様に認められたが、p85を過剰発現することにより、それは著明に阻害された。 以上から、1)Rasを介するシグナルはインスリン刺激による糖輸送活性促進にはほとんど関与していない。2)チロシンリン酸化蛋白と二量体PI3キナーゼを介するシグナルは、インスリンによるGLUT4糖輸送担体のplasma membrane分画への移動を伴った糖輸送活性促進に働いている。3)チロシンリン酸化蛋白と二量体PI3キナーゼを介するシグナル以外に、GLUT4のtranslocationを介さない糖輸送促進へつながる系の存在が示唆された。 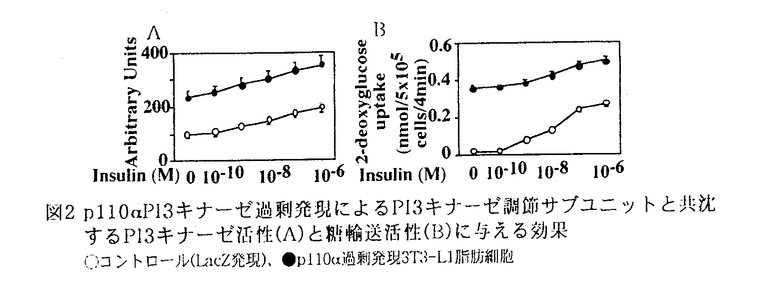 図2p110PI3キナーゼ過剰発現によるPI3キナーゼ調節サブユニットと共沈するPI3キナーゼ活性(A)と糖輸送活性(B)に与える効果○コントロール(LacZ発現)、●p110過剰発現3T3-L1脂肪細胞(2)p110PI3キナーゼの過剰発現 図2p110PI3キナーゼ過剰発現によるPI3キナーゼ調節サブユニットと共沈するPI3キナーゼ活性(A)と糖輸送活性(B)に与える効果○コントロール(LacZ発現)、●p110過剰発現3T3-L1脂肪細胞(2)p110PI3キナーゼの過剰発現 1.p110PI3キナーゼにつけたepitope-tagに対する抗体を用いたimmunoblottingよりp110PI3キナーゼの過剰発現は、確認された。 2.p110PI3キナーゼを過剰発現させた細胞では、インスリン刺激によるチロシンリン酸化蛋白と共沈するPI3キナーゼ活性は、インスリン刺激時、非刺激時共、若干の有意な上昇を認めた。さらに、PI3キナーゼの調節サブユニットと共沈するPI3キナーゼ活性は、インスリン非刺激時にすでにコントロールのインスリン最大刺激時の活性を上回り、また、インスリン刺激により、コントロールのインスリン刺激による活性上昇をも上回るさらなる活性増加が認められた。以上より、少なくともかなりの部分の強制発現させたp110PI3キナーゼは、インスリン非刺激時から細胞内でPI3キナーゼの調節サブユニットと結合して、機能的に発現していることが確認された。 3.2-デオキシグルコース取り込み能は、p110PI3キナーゼを過剰発現させた細胞では、インスリン非刺激時にすでにコントロールのインスリン最大刺激時の取り込み能(非刺激時の12倍)を上回り(コントロール非刺激時の14倍)、インスリン刺激により、さらに1.4倍にまで活性上昇が認められた。 4.GLUT1、GLUT4糖輸送担体の発現は、p110PI3キナーゼを過剰発現させた細胞ではコントロールとはっきりとした差は認めなかった。しかし、遠心分離によるsubcellular fractionation法により、p110PI3キナーゼを過剰発現させた細胞では、インスリン非刺激時にすでにコントロール細胞のインスリン刺激時と同様、plasma membrane分画に多くGLUT1やGLUT4が存在していることが確認された。これは、インスリン添加によってもほとんど変化しなかった。さらに、インスリン非刺激時からGLUT4糖輸送担体が細胞表面に移動していることは、membrane sheet assay法を用いても確認された。 以上から、p110PI3キナーゼの過剰発現によるPI3キナーゼ活性増加により、糖輸送担体蛋白のplasma membraneへのtranslocationを伴う糖輸送能増加がもたらされたことが示唆された。 考察 1.dominant negative Rasの過剰発現により糖輸送担体の発現、インスリンによる細胞内移動、インスリン刺激による糖輸送能に影響がみられなかったことから、3T3-L1脂肪細胞ではインスリンによる糖輸送能促進作用にはMAPキナーゼ系は関与していないことが示唆された。さらに、PI3キナーゼの上流にも下流にもRasを介する系の存在が報告されているが、、これらも脂肪細胞におけるインスリンによる糖輸送能促進作用にはほとんど役割を果たしていないと考えられる。 2.p85の過剰発現の結果より、PI3キナーゼの活性化は、インスリンによる糖輸送担体の細胞内移動に基づく糖輸送能促進作用に必要不可欠な役割を果たしていることが確認された。しかし、PI3キナーゼ活性と糖取り込み活性との間に若干の乖離が認められたことから、チロシンリン酸化蛋白と〓量体PI3キナーゼとの結合を介するシグナル以外に、インスリン刺激により、主に糖輸送担体のintrinsic activityを増加させるシグナル経路が存在することが示唆された。 さらに、p110PI3キナーゼを過剰発現させた細胞で、インスリン刺激により、糖輸送担体のさらなるtranslocationを認めないにもかかわらず、糖輸送活性はさらに増加を示したことから、上記の経路がインスリン添加により刺激された可能性が示唆される。 3.p110PI3キナーゼの過剰発現により、糖輸送担体蛋白のplasma membraneへのtranslocationを伴う糖輸送能増加がもたらされたことから、インスリンの糖輸送能促進作用は、PI3キナーゼの活性上昇にて再現できるものと考えられる。しかし、PDGFなどのインスリン以外の刺激では、量的に同様にPI3キナーゼの活性上昇をもたらしても、インスリンほど著明な糖輸送能促進効果は認められない。これらの結果から、PDGFとインスリンとではPI3キナーゼの活性化が質的に異なるものではないかと考えられる。活性化されたPI3キナーゼの細胞内分布がこれらのシグナル間で異なることが推察されることからも、刺激によるPI3キナーゼ活性の細胞内redistributionが重要である可能性が示唆される。 |