健常な成人では血管の新生はほとんど認められないが、腫瘍組織では増殖に必要な多数の血管が活発に新生していることから、血管内皮細胞に選択的な増殖阻害剤は選択毒性を有すると考えられる。そのため血管新生阻害剤が新しいタイプの抗癌剤として注目されてきたが、効果は腫瘍を退縮させるには至らないのが現状である。また、血管新生阻害剤の効果増強を図った例は過去に無い。そこで本研究では、血管新生阻害剤であるフマギリン誘導体(以下、TNP-470)を用いて、その薬理活性を最大限に引き出し単剤で腫瘍を退縮させ得る実用的な抗腫瘍DDS製剤の設計と最適化を行った。そして実際に担癌動物モデルを用いて抗腫瘍効果の増強効果を検討した。
1.製剤設計と最適化検討1-1.血管新生阻害剤TNP-470のプロフィール TNP-470(図1)はカビ(Aspergillus fumigatus)から産生されるフマギリンの誘導体であり、米国で注射剤としてPhase IIの開発段階にある。TNP-470の水への溶解度は2〜3mg/mlと低く、また、加水分解を受けやすく水溶液中や血漿中では非常に不安定である。一方、有機溶媒や油性基剤へは100mg/ml以上の溶解度で溶解する。生体に投与されたTNP-470は各組織に広く分布し、血液中での分解も含め速やかに代謝され尿および糞中に排泄され、人での全身クリアランスは10〜20l/minと非常に大きい。TNP-470は非常に低いIC50値(数十pg/ml)で血管内皮細胞の増殖を静細胞的に抑制し、高濃度(3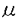 g/ml以上)では殺細胞効果を示す。一方、ほとんどの正常細胞および腫瘍細胞では静細胞的阻害作用がみられず、殺細胞効果が数g/ml以上の濃度で認められる。実際に種々のin vivo腫瘍モデル系において血管内皮細胞増殖阻害に基づく抗腫瘍活性を示し、投与方法を急速静注から点滴静注に変更すると生存日数が大幅に延長し体重減少も緩和されることが報告されている。
g/ml以上)では殺細胞効果を示す。一方、ほとんどの正常細胞および腫瘍細胞では静細胞的阻害作用がみられず、殺細胞効果が数g/ml以上の濃度で認められる。実際に種々のin vivo腫瘍モデル系において血管内皮細胞増殖阻害に基づく抗腫瘍活性を示し、投与方法を急速静注から点滴静注に変更すると生存日数が大幅に延長し体重減少も緩和されることが報告されている。
 図1.TNP-470の化学構造1-2.製剤設計
図1.TNP-470の化学構造1-2.製剤設計 TNP-470の薬理活性を効果的に引き出し低副作用で腫瘍を退縮させることを製剤のコンセプトとし、そのために具備するDDSとして、1)腫瘍部位への薬物ターゲティングと2)薬物徐放を考え製剤設計を行った。抗癌剤のターゲティングには腫瘍部位に親和性を有するキャリヤーの利用が試みられているが、全身投与での効果は十分でない。一方、臨床では腫瘍栄養動脈へ抗癌剤を直接投与する治療が行われている。静脈内投与と動脈内投与のターゲティング効果について、癌患者を想定して構築した生理学的モデルを用いて薬物速度論的に考察すると、静脈内投与と動脈内投与では血中薬物濃度推移に大差は無いが、腫瘍組織中累積薬物量は動脈内投与が大きく上回り、その差は腫瘍組織初回通過時の滞留あるいは取り込み量でほぼ決まると理解できる。また、動脈内投与の場合では腫瘍組織中累積薬物量が全身クリアランスの変動の影響を受けにくく、腫瘍組織への一定薬物量の送達という観点からすれば患者の生理的状態の影響を受けにくい投与方法であると言える。また、TNP-470は急速静注よりも点滴静注で延命効果が優れることから、薬物ターゲティング後にTNP-470が持続的に腫瘍組織を曝露するように薬物徐放が必要である。
1-3.製剤化と最適化検討 動脈内投与後にTNP-470の腫瘍ターゲティングと徐放が期待できるDDS製剤として1)腫瘍組織中の血管塞栓による滞留と主薬徐放を期待する固形のマイクロカプセル製剤、2)腫瘍組織中の微細血管の塞栓ならびに腫瘍血管構造の特殊性による滞留と主薬徐放を期待する油性溶液製剤を考え、薬物徐放性、製剤保存安定性を調べた。マイクロカプセル製剤については、ポリDL-乳酸/グリコール酸共重合体(PLGA)を基剤とした液中乾燥法、または各種固形油性基剤を用いたスプレーチリング法によって主薬封入率がほぼ100%の新規な製剤(粒子径:125〜250m)を数種類調製した。しかしながら、PLGAマイクロカプセルでは薬物放出とは別に製剤中での分解が無視できないことや、他のいずれの製剤も室温での保存安定性が不十分であることが示唆される結果であった。
油性溶液製剤については、種々製剤を調製し薬物徐放性を比較した結果、投与が容易な低粘度のMCT(medium-chain triglyceride、トリ(カプリル/カプリン酸)グリセリン)製剤の徐放性が最も優れ、2週間程度の徐放が可能であることがわかった。また、長期保存安定性も極めて良好であった。なお、MCTはヤシ油を原料とする半合成脂肪酸トリグリセリドで安定な油性基剤であり、経静脈栄養用のエマルジョン製剤としてすでに人に用いられている。
1-4.肝動脈内投与後のTNP-470の組織分布 最も優れた製剤特性を有するMCT製剤の肝臓腫瘍ターゲティング能を調べる目的で、Walker256肝担癌ラットの肝動脈内に製剤を投与し組織分布を調べたところ、MCT製剤は肝臓正常部や他の正常組織に比較して腫瘍組織選択的に高い割合で長期間滞留する優れた腫瘍ターゲティング能を有することがわかった。






