| 【背景】 特発性尿細管性蛋白尿症は1980年に岡田、鈴木らが一つの疾患として提唱し、その後わが国で多数例が報告されている原因不明の疾患である。 本症にもっとも特徴的な検査所見は蛋白尿で、そのうち低分子蛋白の占める割合が高い。また小児期の本症患者の多くは無症状で、3歳時検尿、学校検尿や偶然の検尿にて蛋白尿を指摘され、診断されることが多い。現在、本症は1992年、鈴木らによって提唱された暫定的診断基準に従って診断されることが多い(表1)。 本症例の多くは患者の母親の尿中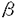 2ミクログロブリンの軽度上昇が認められ、X連鎖性遺伝を強く疑わせる家系が多い。本症の成因は不明であり、特別な治療法はないが各種尿細管機能障害への対症療法が行われることがある。 2ミクログロブリンの軽度上昇が認められ、X連鎖性遺伝を強く疑わせる家系が多い。本症の成因は不明であり、特別な治療法はないが各種尿細管機能障害への対症療法が行われることがある。 一方1964年英国のDentとFriedmanは2例の尿細管機能異常に起因すると推定されるくる病患者を報告した。1990年、WrongらはDentらの報告したこの2例の患者を含む8家系の患者の病態を詳細に検討し、Dent病(高カルシウム尿、尿細管性蛋白尿、くる病、腎石灰化を来たし、最終的に腎不全となる家族性尿細管異常症)と命名した。遺伝様式については初め常染色体遺伝であるとしたが、後にX連鎖性と訂正した。  表1. 特発性尿細管性蛋白尿症の暫定的診断基準 表1. 特発性尿細管性蛋白尿症の暫定的診断基準 1993年、PookらはWrongらの報告した2家系33名の連鎖解析をおこない、Dent病の原因遺伝子の位置がXp11.22に位置することを報告した。またFisherらは同領域にDent病の原因遺伝子と思われる新しいクロールチャンネル遺伝子(CLCN5)を単離同定した。1995年Lloydらは8家系のDent病患者と北アメリカの2家系のX染色体劣性腎結石症患者、イタリアの1家系のX染色体劣性低リン血性くる病患者のCLCN5遺伝子を解析し、すべての症例にCLCN5の異常が存在することを証明した。 1994年五十嵐らはわが国の特発性尿細管性蛋白尿症とDent病の臨床的類似性を指摘し、両者が同一疾患である可能性を指摘した。さらにLloyd、五十嵐らは母親の尿中2ミクログロブリンが高値である特発性尿細管性蛋白尿症4家系のCLCN5遺伝子を解析し、全例にCLCN5遺伝子の異常を証明した。 【目的】 鈴木の診断基準を満たす特発性尿細管性蛋白尿症の中で、臨床的にX連鎖性でない家系や遺伝性が不明の家系が存在する(=母親の尿中2ミクログロブリンが高値でない例、不明な例が存在すること)。すなわち、わが国の特発性尿細管性蛋白尿症は臨床遺伝学的見地からも単一疾患ではない可能性が高い。本研究では鈴木の診断基準を満たす特発性尿細管性蛋白尿症のheterogeneityを細胞遺伝学的に明らかにすることを目的にCLCN5遺伝子の解析を行った。 【対象】 鈴木の診断基準を満たす10家系14名の本症患者を対象とした。その内患児の母の尿中2ミクログロブリン高値(260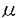 g/l以上)であったのは4家系、正常(260g/l以下)であったのは3家系、不明であったのは3家系である。 g/l以上)であったのは4家系、正常(260g/l以下)であったのは3家系、不明であったのは3家系である。 10名の患者全例に尿中2ミクログロブリンの著明な上昇と尿中カルシウム排泄の増加が認められた。6例に腎エコーあるいはCTにて腎の石灰化が認められた。くる病の罹患者はなく、X線検査にて腎尿路系の奇形はみられなかった。 尚、患者、家族、コントロール全例にインフォームドコンセントを得た。 【方法】 末梢静脈血白血球よりDNAを抽出し、CLCN5遺伝子の11エクソンに13対のプライマーを用いてゲノムのPCRを行った。Taqポリメラーゼを用い、40サイクルの94℃での変性、60℃あるいは55℃(エクソン6、10、11)でのアニーリング、72℃での合成反応を各30秒ずつ行い、2%アガロースゲル(SELECT AGAR;GIBCO BRL)に150Vで電気泳動し、検出した。 1lのPCR産物をPharmacia Phast electrophoresis systemを用いて12.5または20%ポリアクリルアミドゲルで電気泳動を行った。泳動後のゲルは固定した後、銀染色にて変異を検出した。 SSCPで変異が予想されるサンプルについてSequencer(ABI373A)用Sequence mixにてラベルし、Sequencer(ABI373A)をを用いて6%ポリアクリルアミドゲルで電気泳動し、コンピュータにて解析した。尚、SSCPにて変異が見いだせなかった症例はすべてのエクソンについてシークエンスした。 その結果、変異の認められた症例では変異によって出現あるいは消失する制限酵素切断部位を求め、その制限酵素で消化した。また適当な制限酵素切断部位がない症例に対しては正常および患者固有のSequence-specific oligonucleotides(SSO)を用いたドットブロット法によって変異の有無を確認した。 欠失の認められた1家系についてはサザンブロッティング法にてCLCN5遺伝子に隣接するプローブを用いて欠失領域を検討した。 今回の解析により明らかにされたCLCN5遺伝子の変異については、56名の正常日本人(女性54名、男性2名、110アレル)の末梢血より得たコントロールDNAについて変異の有無を検討した。 【結果】 鈴木の診断基準を満たす本症患者10家系中6家系(60%)にCLCN5遺伝子の異常を証明した(表2)。正常コントロールにはこれらの変異は検出されなかった。 家系A、F、GではCLCN5遺伝子のコード領域に変異は認められなかった。家系Jには、CLCN5遺伝子の変異は認められたが、アミノ酸の変異を生じさせないneutral polymorphysmであった。この変異も正常コントロールには認められなかった。  表2;鈴木の診断基準を満たす特発性尿細管性蛋白尿症7家系のCLCN5遺伝子変異【考察】 表2;鈴木の診断基準を満たす特発性尿細管性蛋白尿症7家系のCLCN5遺伝子変異【考察】 CLCN5遺伝子の変異を認めた6家系の特発性尿細管性蛋白尿症のうち、母親の尿中2-microglobulinが高値であったのが4家系、不明であったのが2家系であった。一方、変異のなかった家系では母親の尿中2-microglobulinが高値であったのが1家系、正常が3家系であった。以上の結果より、鈴木らの診断基準をみたす本症の患者では、母親の尿中2-microglobulinが高値である場合はCLCN5遺伝子の転写領域に変異が存在する可能性が極めて高いと考えられた。 本研究およびLloyd,Igarashiらの解析結果とを合わせると、鈴木の診断基準を満たす本症の14家系中10家系(71%)にCLCN5の異常を認めたことになる。またLloyd,Igarashiらの報告の1例と今回検索した1例にみられたコドン279のTGGからTGAに変化する変異はDent病の英国家系と同一の変異であった。一方、今回の解析結果、Lloyd,Igarashiの解析結果及びLloydらのDent病の解析結果から変異のあった21例中19種類の変異が認められ、CLCN5の変異は多様であることが推定できる。さらにDent病の解析結果を総合するとCLCN5異常症の約20%にCLCN5遺伝子の転写領域でCからTへの塩基変化が生じていることが明らかとなった。今後さらに解析を続け症例数を増やすことによりCLCN5遺伝子の人種による変異の特徴、変異の好発部位、変異の種類と臨床症状との関連性などにつき検討したい。 【まとめ】 わが国の特発性尿細管性蛋白尿症の約60%に英国のDent病の責任遺伝子であるCLCN5遺伝子の変異を認めた。この事実は臨床的に単一疾患ではないとされているわが国の特発性尿細管性蛋白尿症が病因遺伝子の解析の点からも単一疾患ではないことを示めすと共に、Dent病すなわちCLCN5異常症が本症の中核的位置を占める疾患であることを意味する。 |