インスリン受容体遺伝子の変異は主として、インスリン抵抗症A型(type A)、leprechaunism(妖精症)、Rabson-Mendenhall症候群などに代表される高度のインスリン抵抗性症候群をきたし、インスリン受容体異常症と呼ばれる。1988年以来、これらのインスリン受容体異常症において実際にインスリン受容体遺伝子変異が同定された。しかし、どのような遺伝子変異やその組み合わせが、インスリン受容体異常症のどのような臨床型や臨床像と対応するのかという肝心な点は殆ど明らかでなかった。本研究では、(1)type A、leprechaunism、Rabson-Mendenhall症候群を含め計六家系の高度インスリン抵抗症患者でインスリン受容体遺伝子変異を同定しその機能的影響を解析した。(2)次に、同定されたインスリン受容体遺伝子変異と臨床型、臨床像との対応について検討した。(3)併せて、一部のインスリン受容体異常症に対してIGF-I治療を試み、一家系についてはインスリン受容体異常症の出生前診断を行った。 対象:高度のインスリン抵抗性があり、インスリン受容体異常症を疑われた六家系を対象にした。これらの家系については国内外の小児科施設から紹介あるいは依頼を受けて検討する機会を得た。症例1.Rabson-Mendenhall症候群(RM-E):6才、男児。高度のインスリン抵抗性糖尿病(空腹時血糖250mg/dl、空腹時IRI400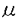 U/ml)。黒色表皮腫、多毛、歯芽早期萌出。症例2.Leprechaunism(大阪):1才、男児。在胎40週2736gにて出生。高度のインスリン抵抗性(空腹時IRI700U/ml)。黒色表皮腫、皮下脂肪萎縮、特異な顔貌。症例3.Leprechaunism(Arab):1才男児。出生時体重1100gと著明な子宮内発育遅延。高度のインスリン抵抗性糖尿病(空腹時血糖446mg/dl、空腹時IRI9175U/ml)。黒色表皮腫、多毛、発育不全、皮下脂肪組織の著明な萎縮、特異な顔貌。両親はいとこ婚であり、共に中等度のインスリン抵抗性を伴うtype Aインスリン抵抗症。症例4.Leprechaunism(埼玉):子宮内発育遅延(出生時体重1296g)、特異な顔貌、皮下脂肪の著明な萎縮、黒色表皮腫。日令12日で高度のインスリン抵抗性糖尿病(空腹時血糖150〜250mg/dl、空腹時IRI2847U/ml)。症例5.Type Aインスリン抵抗症(埼玉):12才、女児。中等度のインスリン抵抗性(空腹時IRI21〜70U/ml)。黒色表皮腫、多毛。症例6.Type Aインスリン抵抗症(鎌ヶ谷):13才、男児。インスリン抵抗性糖尿病(空腹時血糖200mg/dl、空腹時インスリン値149U/ml)。黒色表皮腫。 U/ml)。黒色表皮腫、多毛、歯芽早期萌出。症例2.Leprechaunism(大阪):1才、男児。在胎40週2736gにて出生。高度のインスリン抵抗性(空腹時IRI700U/ml)。黒色表皮腫、皮下脂肪萎縮、特異な顔貌。症例3.Leprechaunism(Arab):1才男児。出生時体重1100gと著明な子宮内発育遅延。高度のインスリン抵抗性糖尿病(空腹時血糖446mg/dl、空腹時IRI9175U/ml)。黒色表皮腫、多毛、発育不全、皮下脂肪組織の著明な萎縮、特異な顔貌。両親はいとこ婚であり、共に中等度のインスリン抵抗性を伴うtype Aインスリン抵抗症。症例4.Leprechaunism(埼玉):子宮内発育遅延(出生時体重1296g)、特異な顔貌、皮下脂肪の著明な萎縮、黒色表皮腫。日令12日で高度のインスリン抵抗性糖尿病(空腹時血糖150〜250mg/dl、空腹時IRI2847U/ml)。症例5.Type Aインスリン抵抗症(埼玉):12才、女児。中等度のインスリン抵抗性(空腹時IRI21〜70U/ml)。黒色表皮腫、多毛。症例6.Type Aインスリン抵抗症(鎌ヶ谷):13才、男児。インスリン抵抗性糖尿病(空腹時血糖200mg/dl、空腹時インスリン値149U/ml)。黒色表皮腫。 方法:これらの六症例について、EBウイルスでトランスフォームした培養リンパ球を用いて、インスリン受容体の、(1)インスリン結合、(2)チロシンキナーゼ活性を検討した。次いで1355アミノ酸からなるインスリン受容体の全ての蛋白コード領域とエクソン・イントロン接合部の全塩基配列をPCR-直接塩基配列決定法により決定した。更に得られた変異インスリン受容体をCHO細胞に発現し、変異受容体の生合成、インスリン結合やチロシンキナーゼ活性、生物学的作用などの機能的検討を行った。また、一部の症例に行った家系調査と併せてインスリン受容体遺伝子変異と臨床型、臨床像との相関を検討した。さらに、一部のインスリン受容体異常症について若干の新しい治療上の試みを行った。 成績 1.インスリン結合は、Rabson-Mendenhall症候群(RM-E)とleprechaunism(大阪)では高度に低下していた。また、leprechaunism(埼玉)、type A(鎌ヶ谷)ではインスリン結合は正常下限より約50%低下していた。一方、leprechaunism(Arab)では、正常であった。インスリン受容体当たりのチロシンキナーゼ活性は、インスリン受容体当たりの活性にして1eprechaunism(Arab)、leprechaunism(埼玉)では約5%と高度に低下していた。Rabson-Mendenhall症候群(RM-E)、leprechaunism(大阪)、type A(鎌ヶ谷)では25〜50%に低下していた。一方、type A(埼玉)では正常であった。 2.これらの六症例で計七種類の遺伝子変異を同定した。症例1.RM-E:イントロン4のスプライス・アクセプター部位のコンセンサス配列AGをGGに変換する点変異とエクソン12の8塩基対欠失の複合ヘテロ接合体であった。いずれも膜挿入部位よりN末端側で欠失をおこし機能的受容体はコードしない。症例2.Leprechaunism(大阪): サブユニット細胞外ドメインのThr(ACG)910→Met(ATG)のミスセンス変異とエクソン19の1塩基対欠失の複合ヘテロ接合体であった。前者は細胞内でのプロセシング障害より細胞膜への挿入が低下し、後者はインスリン受容体mRNA量の著明な低下を起こす。症例3.Leprechaunism(Arab):チロシンキナーゼドメインのArg(CGG)1092→Gln(CAG)のミスセンス変異のホモ接合体であった。この変異受容体はチロシンキナーゼ活性が90%以上低下していた。症例4.Leprechaunism(埼玉):チロシンキナーゼドメインにMet(ATG)1153→Thr(ACG)のミスセンス変異を同定。もう1つの対立遺伝子座にはインスリン受容体遺伝子のmRNA量を著明に低下させる変異の存在が示唆され、複合ヘテロ接合体と考えられた。症例5.Type Aインスリン抵抗症(埼玉):エクソン19に1塩基対の欠失を認め、この変異の単純ヘテロ接合体であった。この一塩基対欠失は症例2で同定されたものと同一であり、インスリン受容体mRNA量の著明な低下をきたした。症例6.Type Aインスリン抵抗症(鎌ヶ谷):チロシンキナーゼドメインのATP結合部位近傍にPro986(CCG)→Leu(CTG)のミスセンス変異を認め、単純ヘテロ接合体と考えられた。 サブユニット細胞外ドメインのThr(ACG)910→Met(ATG)のミスセンス変異とエクソン19の1塩基対欠失の複合ヘテロ接合体であった。前者は細胞内でのプロセシング障害より細胞膜への挿入が低下し、後者はインスリン受容体mRNA量の著明な低下を起こす。症例3.Leprechaunism(Arab):チロシンキナーゼドメインのArg(CGG)1092→Gln(CAG)のミスセンス変異のホモ接合体であった。この変異受容体はチロシンキナーゼ活性が90%以上低下していた。症例4.Leprechaunism(埼玉):チロシンキナーゼドメインにMet(ATG)1153→Thr(ACG)のミスセンス変異を同定。もう1つの対立遺伝子座にはインスリン受容体遺伝子のmRNA量を著明に低下させる変異の存在が示唆され、複合ヘテロ接合体と考えられた。症例5.Type Aインスリン抵抗症(埼玉):エクソン19に1塩基対の欠失を認め、この変異の単純ヘテロ接合体であった。この一塩基対欠失は症例2で同定されたものと同一であり、インスリン受容体mRNA量の著明な低下をきたした。症例6.Type Aインスリン抵抗症(鎌ヶ谷):チロシンキナーゼドメインのATP結合部位近傍にPro986(CCG)→Leu(CTG)のミスセンス変異を認め、単純ヘテロ接合体と考えられた。 3.症例3のleprechaunism(Arab)に対して、IGF-1治療を行った。インスリン受容体チロシンキナーゼ活性の著明な低下を認める本症例において、IGF-Iは糖尿病の改善とともに、成長・発達遅滞の改善に有効であった。患児の培養皮膚線維芽細胞において、インスリンによっては促進されなかったIRS-1のチロシンリン酸化、糖取り込みはIGF-1により両者とも健常対照と同程度に促進された。 4.Leprechaunism(Arab)の家系では第1子をすでに同症で死亡させており、両親の強い希望とインフォームド・コンセントのもとに第5子の胎児診断を施行した。胎児診断は、胎生17週に羊水胎児細胞より抽出したDNAを用いて、本変異(Arg1092-Gln)により制限酵素PstIの認識部位が新たに1カ所生じることを利用して行った。この結果から胎児はヘテロ接合体と考えられ、leprechaunismは発症しないと考え妊娠を継続した。実際新生児はヘテロであり、leprechaunismを呈さなかった。インスリン受容体異常症の家系において出生前診断が正確であった。 考察と結論 本研究で検討した六家系について、臨床的に重症度の高いleprechaunismやRabson-Mendenhall症候群では、2つのalleleの両方に変異を認めるホモ接合体や複合ヘテロ接合体であったのに対し、この2症候群に比し、やや重症度の低いtype Aは1つのalleleにのみ変異を認めるヘテロ接合体であった。特に、血縁関係のないleprechaunism(大阪)とtype A(埼玉)に共通のインスリン受容体遺伝子変異(コドン1109の一塩基対欠失によるフレームシフト変異)を同定したが、この変異ともう1つのalleleのミスセンス変異(Thr910→Met)の複合ヘテロはleprechaunismを呈し、一方、フレームシフト変異の単純ヘテロはtype Aを呈した。更に、症例3のleprechaunism(Arab)はArg1092→Glnのミスセンス変異のホモであったが、この変異のヘテロである両親はtype Aであった。これらの知見より、インスリン受容体異常症の臨床型や重症度は、インスリン受容体遺伝子変異の重症度や総チロシンキナーゼ活性によって規定されること、従来異なる疾患と考えられていたleprechaunismとtype Aインスリン抵抗症はインスリン受容体遺伝子異常の重症型と軽症型という一つの連続したスペクトラムの2病型としてとらえられることが明らかとなった。 |