薬物は標的生体高分子に結合することで、その生理活性を発現することが知られている。したがって、酵素などの受容体の立体構造が解明されてる場合は、その受容体に対して特異的に結合可能な化合物をコンピュータで選択または設計できれば、新規阻害剤の創出に大きく貢献できるはずである。さらに、近年のX線やNMRによる三次元タンパク構造解明の加速化、コンピュータの高性能化、ドッキング計算のプログラム開発により、Structure Based Drug Design(SBDD)を行う基盤が整えられてきた。この方法では、立体構造が解明されている受容体に対して活性未知の化合物をドッキングして、その複合体の安定性を評価することで合成やアッセイする化合物を選択する。この計算で大きな問題となるのは、回転可能結合(フレキシブル結合)による非常に多くのコンフォメーションが、ドッキング対象化合物にとって可能な点である。ドッキング計算で構造的自由度を考慮するには多大な計算時間と適切な計算アルゴリズムが必要であるため、初期のドッキング計算では、この自由度を完全に無視した計算が行われていた。その後、構造的自由度を無視したドッキング計算でHIVプロテアーゼに阻害活性がある化合物が発見されて注目を集めたが、データベース中の化合物の初期コンフォメーションが受容体と複合体形成できる可能性は極めて低いので、ドッキング計算の精度向上には化合物の構造的自由度の考慮が必然である。しかし、化合物のフレキシブル結合数が多いとコンフォメーション空間が莫大になるので、構造的自由度を考慮したドッキング計算を大規模化合物データベースに適用するのは非常に困難である。したがって、ドッキング計算方法の大幅な効率化が望まれる。 まず、構造的自由度を考慮した自動ドッキング計算のプログラムを構築した。本研究のドッキングにおけるコンフォメーション探索では、化合物中の構造的自由度を有さないフラグメントをドッキングした後、順次フラグメントを付加して受容体中でコンフォメーション探索を行う方法(階層的コンフォメーション探索法)を採用した。この階層的コンフォメーション探索法では、受容体構造によりドッキング対象化合物のコンフォメーション空間が制限されるので、化合物のコンフォメーションを発生させてから、受容体にドッキングする方法に比較して効率がよいと考えた。また、ドッキング対象化合物を特別な水素原子タイプでプロトン化した後、局部的相互作用に基づいてその水素原子の有無を変化させることにより、化合物の複数プロトン化状態を同時に考慮してドッキング計算する方法を開発した。(図1)  図1 化合物の複数プロトン化状態を同時に考慮したドッキングと階層的コンフォメーション探索 図1 化合物の複数プロトン化状態を同時に考慮したドッキングと階層的コンフォメーション探索 開発したプログラムを用いてACD15068化合物をジヒドロ葉酸還元酵素に対してドッキングして得られた上位36個の化合物を示したが、既知阻害剤(MTX)とその誘導体の多くが安定複合体を形成可能だと判断されて選択されているのがわかる。(図2)  図2 ACD15068化合物のジヒドロ葉酸還元酵素に対するドッキング結果 図2 ACD15068化合物のジヒドロ葉酸還元酵素に対するドッキング結果 次に、仮想化合物ライブラリー生成とその効率的なドッキング計算を行うプログラムの開発を行った。これまで、受容体構造から阻害活性を有すると思われる仮想化合物を自動発生するプログラム(DENOVOプログラム)について数々の報告がなされてきたが、ほとんどの報告では有機合成ルートを考慮せずに仮想化合物を生成しているので、シュミレーションで有望な化合物を見付けても、それを実際に合成することは非常に困難であった。また、アミド結合構築が可能なプログラムも存在したが、合成できる化合物の多様性は極めて限定され、「薬らしさ」を欠如する可能性が高いと考えられる。以上の問題を回避するために、アミド化縮合反応に限定しない仮想有機化学反応により化合物ライブラリーを生成するプログラムを開発した。しかし、仮想反応により生成される化合物ライブラリーを階層的にコンフォメーション探索すると、反応基質由来の共通部分構造を繰り返しドッキングすることになるので、計算効率の点で好ましくない。そこで、反応基質のドッキング計算を行った後、そのコンフォメーション情報を仮想反応の過程で反応生成物に継承させることで、反応生成物の共通部分構造を同時にドッキングできるようにした。(図3)  図3 仮想有機化学反応とコンフォメーション継承 図3 仮想有機化学反応とコンフォメーション継承 また、安定複合体を形成できない反応中間体を効率的に除去できることも、仮想反応とドッキング計算を順番に行うことの利点である。(図4) 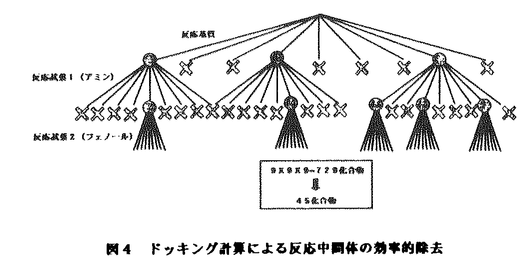 図4 ドッキング計算による反応中間体の効率的除去 図4 ドッキング計算による反応中間体の効率的除去 さらに、この共通部分構造の同時ドッキング方法の概念を拡張し、反応生成物の類似部分構造を考慮してコンフォメーション探索の対象空間を限定することでコンフォメーション探素を効率化するプログラムの開発を行った。このプログラムは、ドッキング対象化合物をクラスター化した後,各クラスターの代表化合物のドッキング計算により得られるコンフォメーションにより、クラスター中の個別化合物のコンフォメーション探索空間を制限することでドッキング計算を効率化する。(図5)この計算方法を使用したテストにで、計算時間が5倍以上に短縮できることが判明した。  図5 プログラムELECT++の化合物類似性によるコンフォメーション探索効率化法 図5 プログラムELECT++の化合物類似性によるコンフォメーション探索効率化法 また、真空中のドッキング計算では考慮されない溶媒効果を複合体の安定性評価に反映させるために、複合体形成時に解放される水分子によるエントロピー増加量を近似する評価関数の開発についても行った。まず、レセプター内部を20方向に伸張したベクトルにより定義した後、レセプター内部のリガンド体積を計測して水分子によるエントロピー増加に比例量だと考えた。(図6)この評価方法を使用して、ドッキング結果のコンフォメーションを選択したところ、計算結果と阻害活性値に高い相関を見出すことができた。  図6 解放される水分子によるエントロピー増加量を近似する評価関数 図6 解放される水分子によるエントロピー増加量を近似する評価関数 本研究では、これまでのドッキング計算の分野で頻繁に行われてきた既存化合物ライブラリーのスクリーニングや、少数の合成困難な化合物の設計を主目的とするのではなく、特に、コンビナトリアルライブラリーの設計に重点を置いてプログラムを開発した。今後は、これらプログラムによる化合物ライブラリーの設計を通じて創薬効率化に貢献したいと考えている。 |